Highlights
Biogen has developed an experimental drug tofersen to treat amyotrophic lateral sclerosis (ALS) with superoxide dismutase 1 (SOD1) gene mutation.
Clinical validation showed that tofersen administration failed to provide an exit to the primary efficacy endpoint of therapy as stated by improvement on the Amyotrophic Lateral Sclerosis Functionality Rating Scale–Revised (ALSFRS-R). However, certain trends were observed in favor of tofersen when it comes to secondary endpoints assessing biological markers and clinical function.
Subsequent integrative analysis of the data allowed to conclude that earlier initiation of tofersen leads to containment of ALS progression: Slowing of deterioration of motor and respiratory functions, muscle strength and quality of life in patients with ALS and SOD1 mutation was noted.
In light of the critical unmet need for amyotrophic lateral sclerosis drugs, Biogen has expanded eligibility for the current Early Access Program (EAP) to all patients with SOD1-ALS living in countries where such programs are legally allowed or may be allowed in the future. The EAP allows patients free access to the drug until it is commercially licensed. It should be understood that the way forward for tofersen development is unclear, and if regulators require Biogen to conduct another controlled trial, the early access program may be revised or closed.
Despite inconsistent clinical results, Biogen notified in late July 2022 that the U.S. Food and Drug Administration (FDA) had accepted the New Drug Application (NDA) for tofersen. The verdict of the U.S. regulator, which is reviewing Biogen’s NDA on an expedited basis, will be rendered by the end of January 2023.
If tofersen passes regulatory scrutiny, it will go on sale under the supposed trademark Qalsody.

In mid-June 2022, Canada became the first country in the world to approve Albrioza (sodium phenylbutyrate + ursodoxicoltaurine), a new drug indicated to treat amyotrophic lateral sclerosis. The oral Albrioza, developed by Amylyx Pharmaceuticals under the codename AMX0035 and implemented in a powder formulation for suspensions, aims to inhibit motor neuron dysfunction and death.

Albrioza: New Drug for Amyotrophic Lateral Sclerosis Treatment
Amylyx Pharmaceuticals proposed mixture of sodium phenylbutyrate and tauroursodeoxycholic acid for ALS treatment.
What Is Amyotrophic Lateral Sclerosis With SOD1 Mutation?
Amyotrophic lateral sclerosis (ALS) is a rare, progressive and fatal neurodegenerative disease that results in the loss of motor neurons in the brain and spinal cord responsible for the control of voluntary muscle contractions. Patients experience muscle weakness and atrophy, which leads to a loss of independence, they gradually lose the ability to move, speak, eat, and eventually breathe. Life expectancy for people with ALS is 3–5 years from the onset of symptoms.
The exact cause of ALS is unknown, it may be either genetic mutations or environmental factors. Mutations in the gene encoding superoxide dismutase 1 (SOD1) are known to be responsible for about 2% of all ALS cases and 20% of familial ALS cases.
The enzyme superoxide dismutase 1 is a powerful antioxidant that protects the body from damage caused by superoxide, a toxic free radical produced in the mitochondria. Free radicals are highly reactive molecules produced in cells during normal metabolism. Free radicals can cause damage to DNA and proteins in cells. It is suggested that aggregate accumulation of mutant SOD1 may play a role in cellular dysfunction by damaging mitochondria, proteasomes, protein folding chaperones, and other proteins.
Qalsody: Mechanism of Action of Tofersen
Tofersen (BIIB067, ISIS-SOD1Rx) is a second-generation antisense oligonucleotide (ASO) that targets the degradation of superoxide dismutase 1 (SOD1) mRNA to prevent SOD1 protein synthesis and a corresponding reduction in its levels.
Mutant SOD1 is thought to have a toxic effect on motor neurons, and therefore reducing its level may have therapeutic value during the treatment of SOD1-mutant amyotrophic lateral sclerosis (ALS). Overexpression of mutant SOD1 in mice or rats replicates important aspects of ALS, including loss of neuromuscular junction innervation and motor neuron death. [1] [2] Loss of SOD1, although it ultimately leads to motor neuron dysfunction, it still does not lead to motor neuron death. [3] [4] Individual mutations in ALS are associated with different levels of SOD1 activity, but there is no correlation between disease severity and SOD1 activity. [5] [6] [7]
In 2013, Ionis Pharmaceuticals, the company behind tofersen development, tested ISIS 333611, the first generation of ASO against SOD1, confirming the safety of the experimental therapy. [8]
Then Ionis, which carried out an in vitro screening of more than two thousand ASOs, identified two molecules targeting the 3′-untranslated region of SOD1 mRNA, the blocking of which resulted in maximum effectiveness in the task of reducing the expression and level of SOD1 protein in cells. In animal models, it has been shown that single injections of the appropriate drug compounds into the brain or spinal cord lead to a delay in ALS manifestation, improve the function of motor neurons, and prolong survival. Preservation of neuromuscular innervation and control of serum phosphorylated neurofilament heavy chain levels, a prognostic biomarker of ALS, have been observed. [9]
In the NCT02623699 phase 1/2 clinical trial, the mechanism of action of tofersen was confirmed, manifested by a decrease in the level of SOD1 in the cerebrospinal fluid. [10]
In December 2018, Biogen licensed tofersen from Ionis for $35 million upfront, with subsequent payments of up to $55 million as the project develops, plus royalties from sales of the finished drug.
Qalsody: Efficacy and Safety of Tofersen in SOD1-ALS Treatment
The VALOR (NCT02623699, part C) phase 3 (randomized, double-blind, placebo-controlled, multicenter, international) clinical trial enrolled patients (n=108) with amyotrophic lateral sclerosis (ALS) and a confirmed superoxide dismutase 1 (SOD1) gene mutation.
Participants were treated intrathecally with placebo or tofersen at a dose of 100 mg, the first 3 doses every 2 weeks and then 5 more doses every 4 weeks. Concomitant administration of riluzole was allowed, edaravone was not.
The primary endpoint was the change in the score on the Amyotrophic Lateral Sclerosis Functionality Rating Scale–Revised (ALSFRS-R) after 28 weeks of treatment. This change reflects the rate of ALS progression.
Primary data analysis (in a population with fast-progressing disease) revealed no statistically significant difference between the tofersen and placebo groups (difference 1.2 points, p=0.97).
- Fast progression of amyotrophic lateral sclerosis suggested a ≥ 0.2 point per month decline in ALSFRS-R score (for the SOD1 mutation historically associated with reduced survival to less than 3 years) or ≥ 0.9 points per month (for other SOD1 mutations).
Nevertheless, trends were noted in favor of Qalsody on a number of secondary and exploratory indicators that assessed the biological activity of tofersen and clinical functions of patients, including motor function, respiratory function, and quality of life.
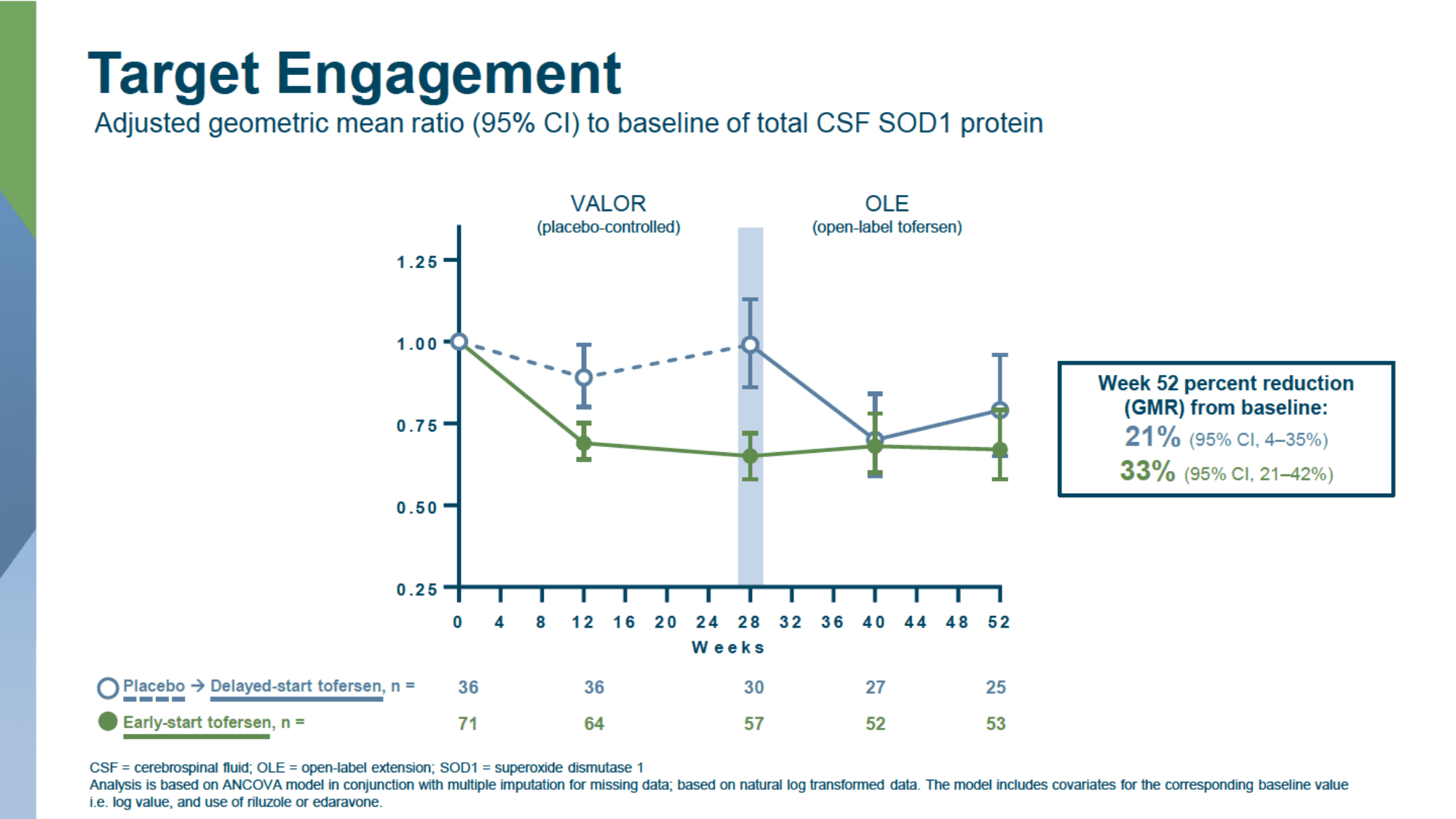
Thus, the difference in change in total cerebrospinal fluid SOD1 protein levels, as a marker of therapeutic target engagement, between the tofersen and placebo groups was 38% and 26%, respectively, in the populations with fast- and slow-progressing ALS (p<0.0001 and p=0.0007).
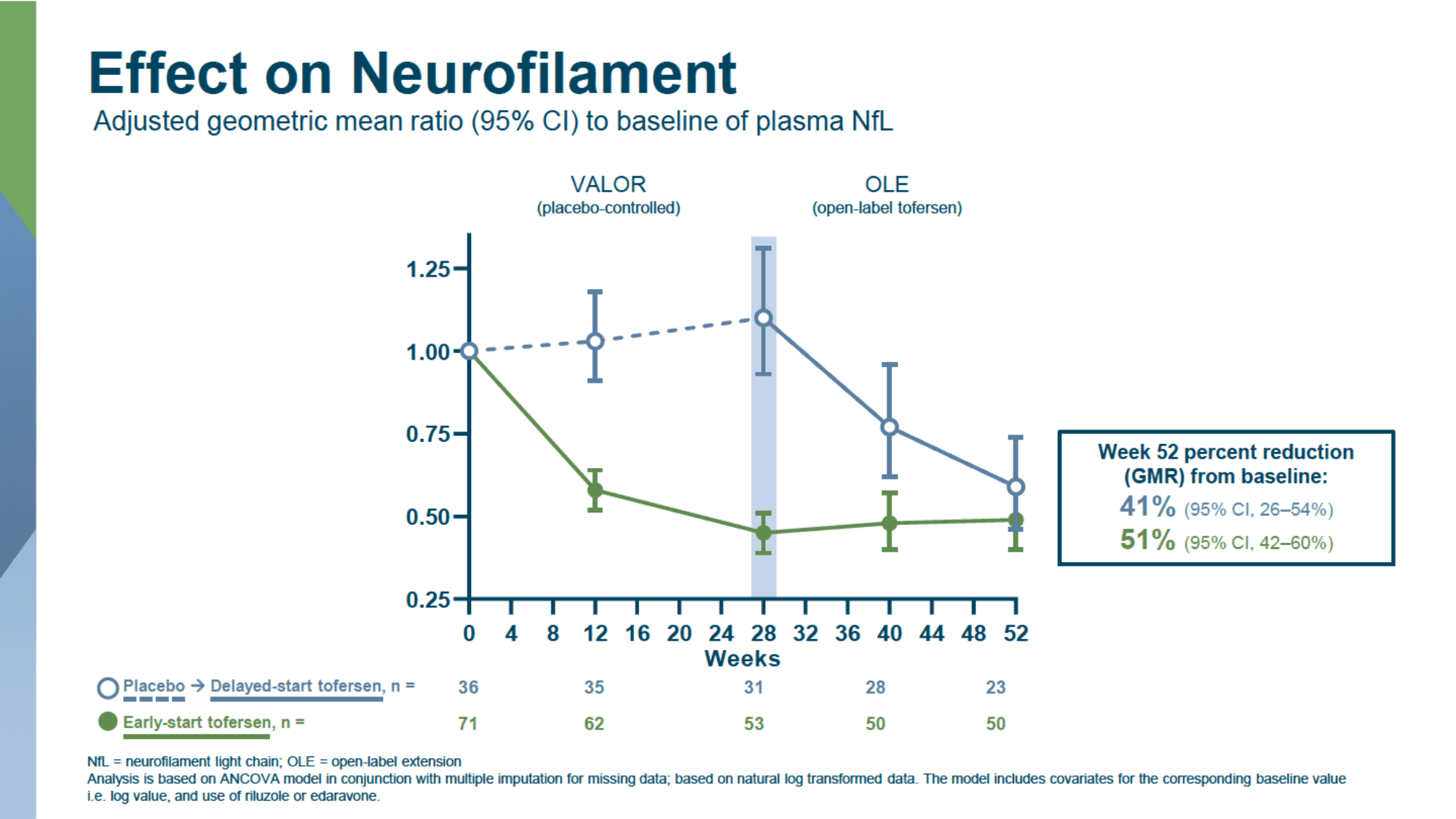
The difference in the change in the plasma concentration of neurofilament light chain (NfL), as a marker of axonal damage and neurodegeneration, between the tofersen and placebo groups was 67% and 48%, respectively, in populations with fast- and slow-progressing ALS.
In the population of fast-progressing ALS, administration of Qalsody provided a slowing of the deterioration of the following indicators (average difference relative to placebo):
- respiratory function, according to the predicted slow vital capacity (SVC): 7,9% (p=0,32)
- muscle strength, according to the handheld dynamometry (HDD) megascore: 0.02 (p=0.84)
- quality of life, according to the Amyotrophic Lateral Sclerosis Assessment Questionnaire (ALSAQ-5): −5.6 points.
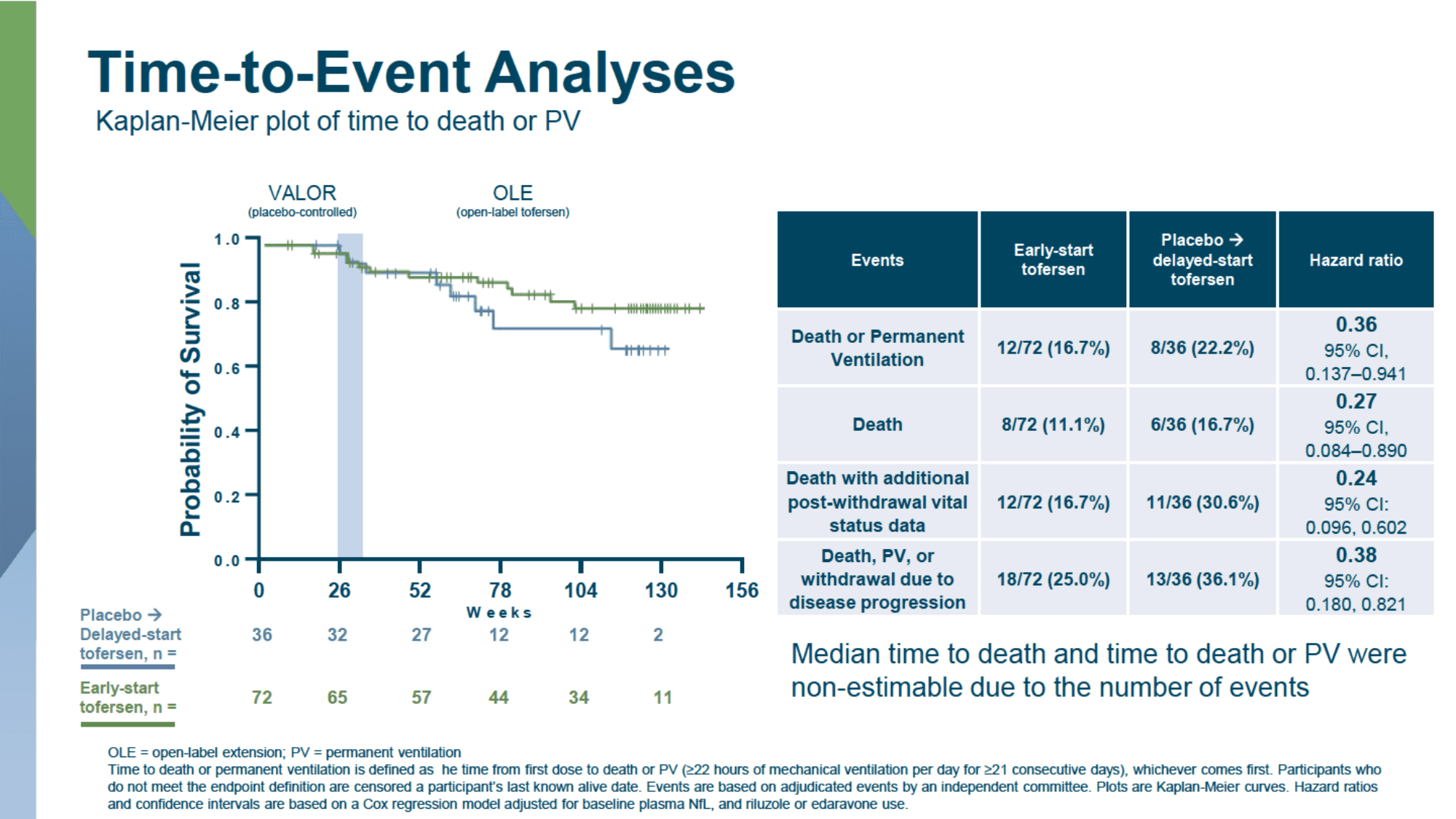
The median time to death or need for permanent ventilation (PM) could not be estimated because of the small number of relevant events over a 28-week period.
Among the most common adverse events (AEs) in patients treated with Qalsody: pain due to intrathecal injection, headache, limb pain, falls, back pain. All were characterized by mild-to-moderate severity. Serious AEs were reported in 18% of patients in the tofersen group and in 14% of patients in the placebo group. Serious neurological events were reported in 5% of those receiving tofersen, including 2 cases of myelitis (2%). In the tofersen group, 6% of patients discontinued participation in the study.
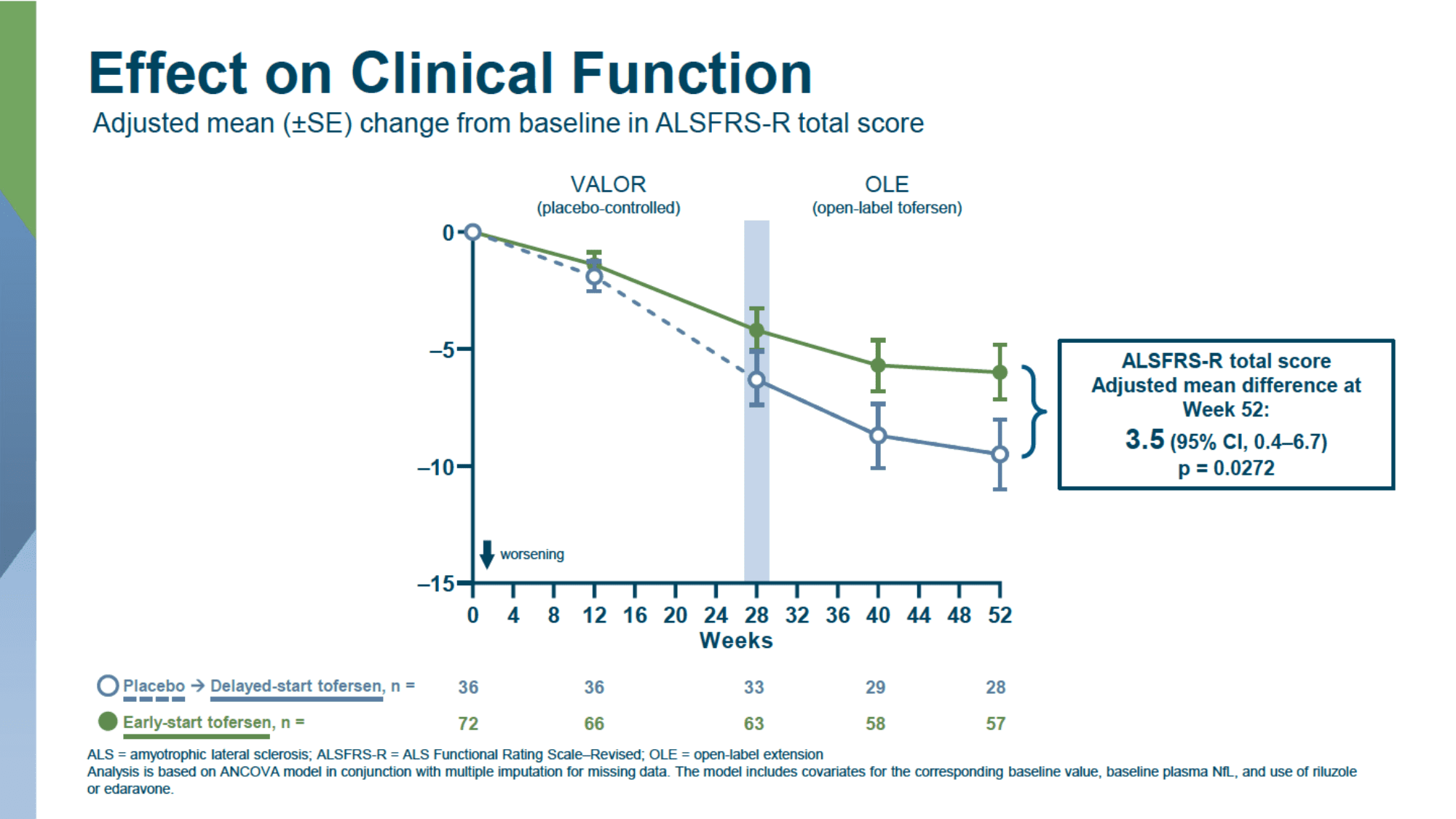
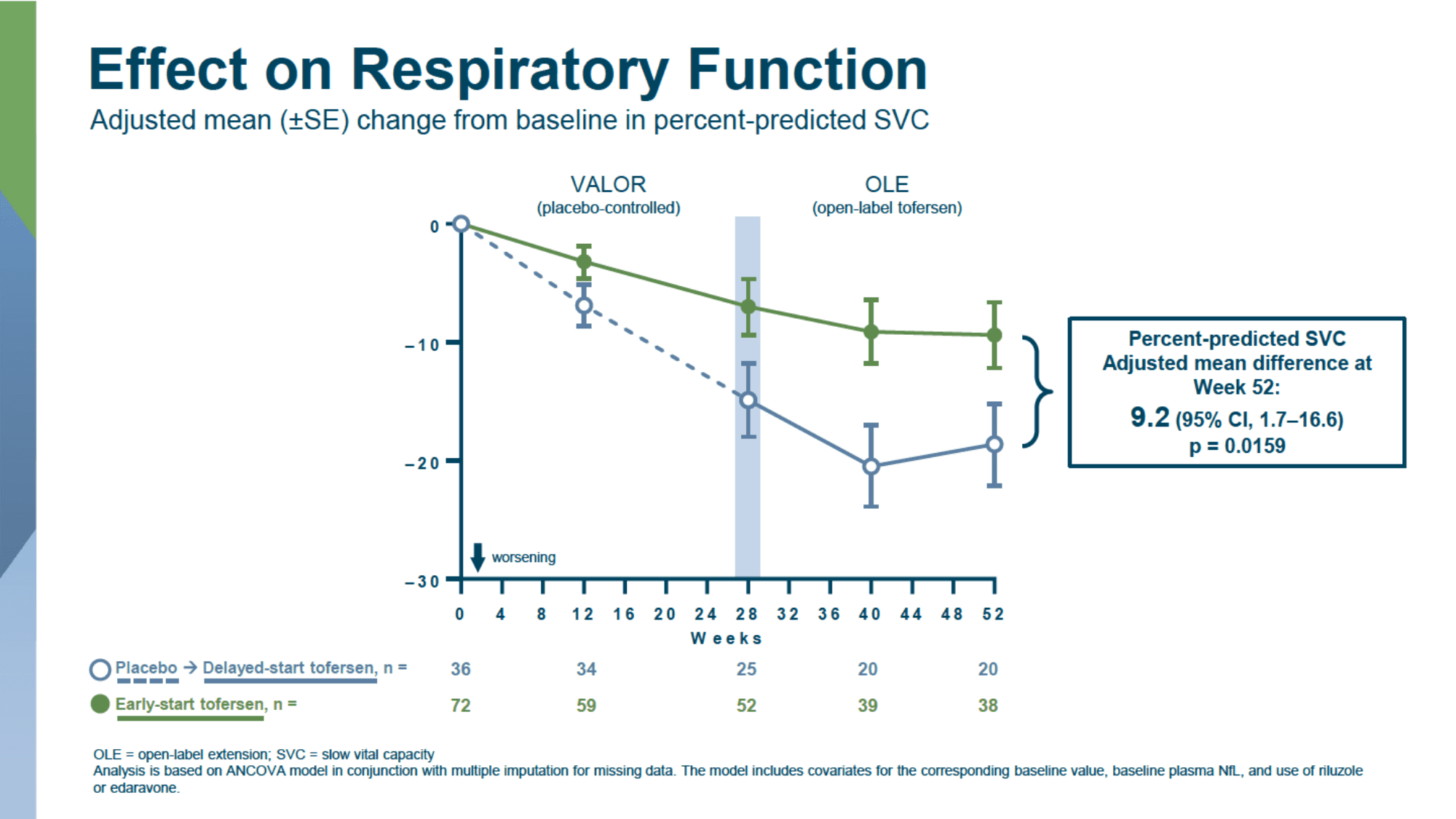
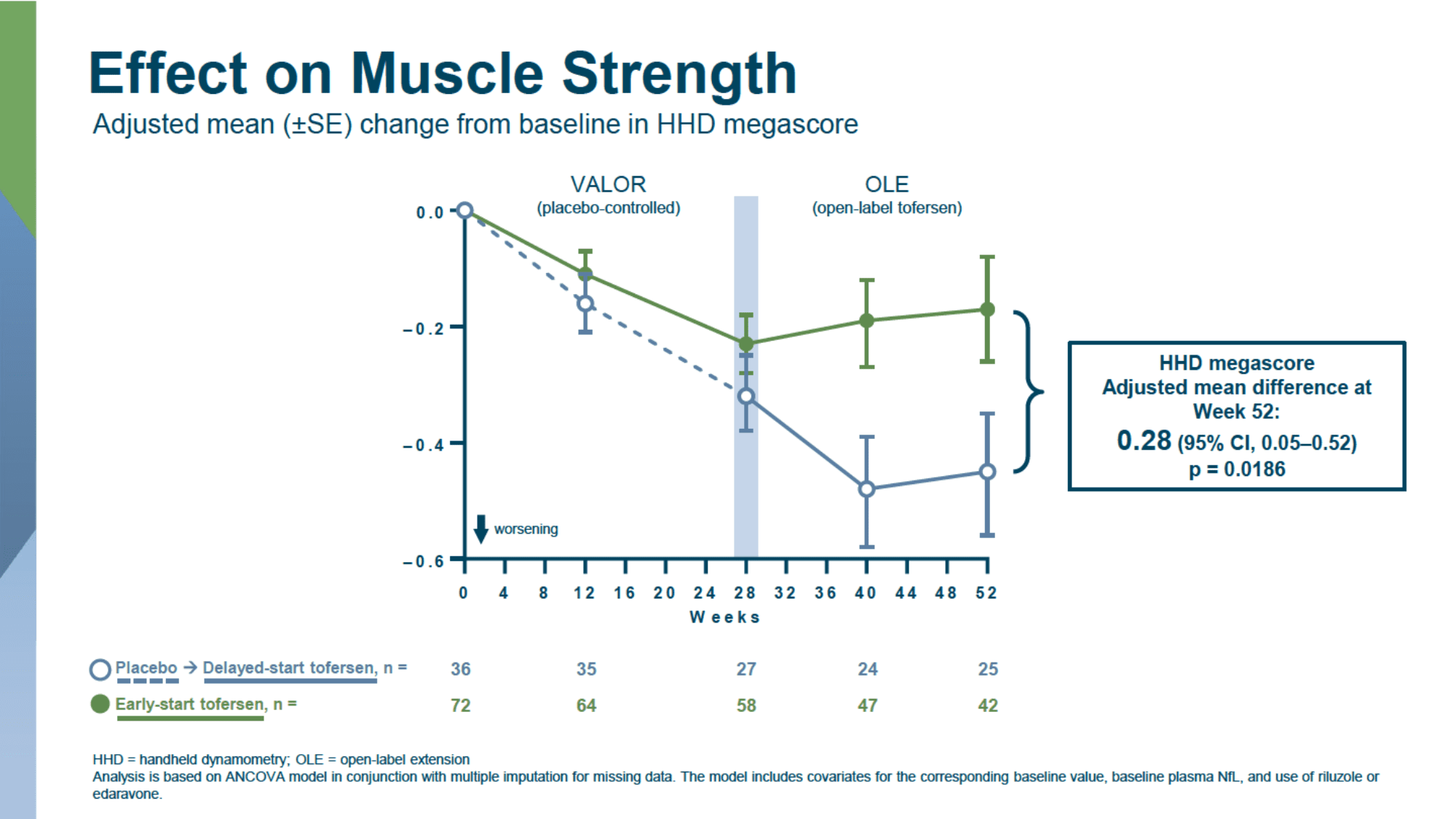
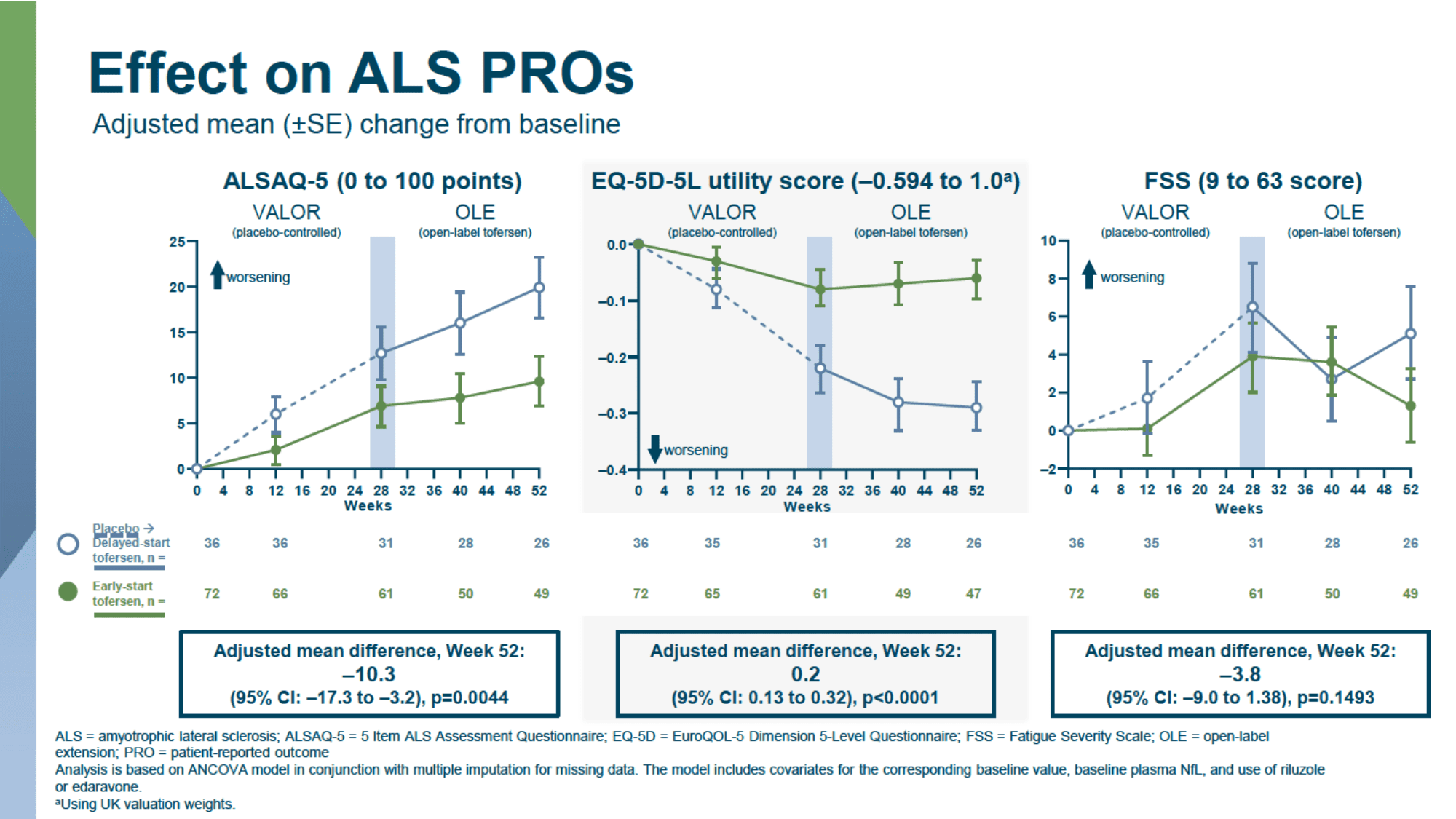
In a longer follow-up of patients who agreed to continue participating in the open-label extension (OLE) of the VALOR study, where all patients were treated with tofersen, it was found that earlier initiation of treatment with the drug consistently slowed clinical deterioration in the entire patient population when compared to delayed use due to the initial receipt of a placebo.
If we compare the results after 12 months of Qalsody administration with its use for 6 months, we found the following mean difference in changes in clinical parameters:
- slowing of progression of amyotrophic lateral sclerosis, according to ALSFRS-R: 3.5 points (95% CI: 0.4 to 6.7, p=0.03)
- delayed progression of worsening respiratory function, according to predicted SVC: 9.2% (95% CI: 1.7 to 16.6; p=0.02)
- slowing the progression of weakening muscle strength, according to HDD megascore: 0.28 (95% CI: 0.05 to 0.52; p=0.02)
- slowing deterioration in quality of life, according to ALSAQ-5: −10.3 points (95% CI: −17.3 to −3.2; p=0.004).
A 12-month therapy of ALS with tofersen confirmed the preservation of the reduced levels of SOD1 and NfL achieved after 6 months of therapy.
By the time the data were analyzed, most participants were alive without the need for permanent ventilation, and therefore the median time to death or transition to PM could not be estimated. However, preliminary data suggest that earlier initiation of Qalsody reduced the cumulative risk of death and need for PM by 64% and the risk of death by 73%: respective hazard ratio (HR) of 0.36 (95% CI: 0.137 to 0.941) and 0.27 (95% CI: 0.084 to 0.890).
In parallel, tofersen is being studied in the ATLAS (NCT04856982) phase 3 (randomized, double-blind, placebo-controlled, multicenter, international) clinical trial, which is testing the suitability of the drug for delaying the clinical manifestation of ALS among presymptomatic adult SOD1 mutation carriers with elevated plasma NfL levels.
Expert Comments
Tofersen’s future looks murky. For Biogen, it’s not such a big deal, because the 2% eligible population of patients with amyotrophic lateral sclerosis and the SOD1 mutation has never been perceived as commercially large-scale. But for Ionis, the therapeutic failure of tofersen is a serious blow to the modality of antisense oligonucleotides. Especially after the failure of tominersen (IONIS-HTTRx), a joint experimental drug with Roche to treat Huntington’s disease; however, the clinical study did not stop. That’s why after the key results of the clinical trial of tofersen were announced, Ionis stock quotes lost 12% at once.
Qalsody failed to reach the primary endpoint assessed by the ALSFRS-R change, the 1.2-point difference with placebo among fast-progressing patients was not statistically significant. In the group of slow-progressing patients, the difference was 1.4 points. At the same time, in the latter population during the ongoing OLE of the VALOR study, tofersen still provided a difference of 2.9 points. According to expert opinion, a difference of 2.0 points can be considered clinically significant.
If we talk about glimmers of hope, there are some. To begin with, it should be understood that tofersen did what it was designed to do: it lowered SOD1 levels, which theoretically should slow the progression of ALS. Now the question arises why this was not reflected in a significant improvement in the clinical parameters of the patients. There is an opinion that the lack of a proper correlation is due, first, to the moderate magnitude of SOD1 reduction, and, second, to the short duration of the trial. Again, tofersen showed a decent decrease in NfL levels, indicating that neurodegeneration was arrested.
Although there were strong doubts that Biogen would risk spending money on a tofersen’s NDA with the current data, the submission was nevertheless made. Most likely, the data collected during the study’s OLE were taken into account.
Even if the FDA experts deny Biogen, it will still wait for the results of the clinical trial of Qalsody in presymptomatic patients: as in many other neurological diseases, toxic protein aggregates can form long before symptoms appear. We’ll have to be patient for a long time, the ATLAS study is scheduled to be completed in the summer of 2026.
In late March 2022, Biogen and Ionis abandoned development of experimental tadnersen (BIIB078, IONIS-C9Rx), an antisense oligonucleotide therapy for ALS with C9orf72 gene mutation. In the NCT03626012 phase 1 clinical trial, intrathecally administered tadnersen failed to demonstrate adequate benefit along with a challenging safety profile.
Ionis is independently pursuing the antisense oligonucleotide ulefnersen (ION363, jacifusen) in the NCT04768972 phase 3 clinical trial in the treatment of ALS with FUS gene mutation.
Extras
Results from the phase 3 VALOR study and its open-label extension: Evaluating the clinical efficacy and safety of tofersen in adults with ALS and confirmed SOD1 mutation. [PDF]
Evaluating efficacy and safety of tofersen in adults with SOD1-ALS: Results from the phase 3 VALOR trial and open-label extension. [PDF]
ALS antisense drug falters in phase III. Nat Rev Drug Discov. 2021 Dec;20(12):883-885. [source]
Phase 1–2 trial of antisense oligonucleotide tofersen for SOD1 ALS. N Engl J Med. 2020 Jul 9;383(2):109-119. [source]
Design of a randomized, placebo-controlled, phase 3 trial of tofersen initiated in clinically presymptomatic SOD1 variant carriers: the ATLAS study. Neurotherapeutics. 2022 May 18.[source]