Highlights
Relyvrio/Albrioza is a new drug for the treatment of amyotrophic lateral sclerosis.
The oral Relyvrio/Albrioza, developed by Amylyx Pharmaceuticals under the codename AMX0035 and formulated in a suspension powder, aims to inhibit motor neuron dysfunction and death.
Canada became the first country in the world to approve Albrioza (sodium phenylbutyrate + ursodoxicoltaurine) in mid-June 2022.
In late September 2022, the U.S. Food and Drug Administration (FDA) approved Relyvrio (sodium phenylbutyrate + taurursodiol).
Amyotrophic lateral sclerosis (ALS) is a relentlessly progressive and fatal neurodegenerative disease caused by the death of motor neurons in the brain and spinal cord. The inexorably worsening course of the disease leads to the weakening of the muscular system, inability to move and speak, paralysis of breathing, and ultimately death.
Existing medical interventions for ALS are mainly symptomatic therapies for complications of amyotrophic lateral sclerosis. Specific drugs, such as riluzole and edaravone, work with very modest efficacy, only slightly limiting the progression of pathology.
Relyvrio/Albrioza reduces the risk of death by 44%. Considering that the median survival rate for ALS is approximately 4 years from symptom onset, continuous administration of Relyvrio/Albrioza can extend life to 6–6.5 years.
That’s not to say that Amylyx has created something incredibly powerful from a healing viewpoint. Still, the emergence of a new drug for ALS is a major event for the patient and physician community.
USA
In early November 2021, a New Drug Application (NDA) for Relyvrio was submitted to the U.S. Food and Drug Administration (FDA).
At the end of March 2022, the FDA advisory committee that reviewed Relyvrio’s NDA was divided, with 4 experts in favor of approving the drug and 6 against. The FDA takes into account the opinions of the invited experts but does not always follow their recommendations. For example, at one time the experts were strongly against approving the highly controversial drug Aduhelm (aducanumab) for treating Alzheimer’s disease, but the American regulator still allowed the drug. In any case, the FDA decision on Relyvrio was expected by the end of June 2022.
In early June 2022, it became known that the FDA has extended the deadline for reviewing the NDA for Relyvrio. The regulator will now have until the end of September 2022 to issue its decision. It is stated that the FDA needs more time to analyze additional data from clinical trials of the drug.
In early September 2022, during the second meeting of the FDA advisory committee, the expert vote was divided: 7 voted for approval of Relyvrio and 2 voted against. There was a mandatory condition: If the drug fails to demonstrate therapeutic efficacy in the ongoing 48-week PHOENIX (NCT05021536) phase 3 clinical trial, which will end by mid- to late-2024, Relyvrio will withdraw from the market.
Experts changed their anger for mercy on Relyvrio after reviewing additional clinical data showing that the drug has the potential to prolong life and listening to the opinions of doctors and patients. Nevertheless, the regulator stayed true to its previous position that the evidence collected was still insufficient to show unequivocally that amyotrophic lateral sclerosis treatment is effective in slowing its progression. The data provided by Amylyx lack unequivocal independence, reliability, and conclusiveness, and, being built only on modified statistical analyses of previous data, are not new in any way. However, since the deadly neurodegenerative disease has a catastrophic lack of drugs, it would be unethical to deny patients access to Relyvrio. The regulator emphasized that it would exercise “the broadest flexibility” in deciding whether to approve the drug, adding that patients and physicians would assume all risks associated with the safety of treatment.
In late September 2022, the U.S. Food and Drug Administration (FDA) approved Relyvrio.

Relyvrio, which is an oral suspension, is mixed in a cup of water at room temperature and taken orally or via feeding tube. Relyvrio is administered before a snack or meal, one packet per day for the first 3 weeks of treatment and then two packets daily.
- Relyvrio (sodium phenylbutyrate + taurursodiol). Prescribing information. U.S. [PDF]
For uninsured American patients, the price of an annual course of treatment with Relyvrio is $158,000.
Relyvrio will be available in the U.S. in the fall of 2022.
While the FDA was nervously considering whether or not to approve Relyvrio, patients in need were looking for ways to buy it in Canada if a Canadian doctor had written a prescription for it. The U.S. generally prohibits the importation of drugs for personal use, but there are a number of exceptions to this rule. For example, it is not illegal to import drugs for a serious disease for which there is no FDA-approved effective treatment, so Albrioza could be imported legally.
In mid-November 2020, I AM ALS, the amyotrophic lateral sclerosis patient community, and The ALS Association, a nonprofit organization that focuses on ALS issues and concerns, as well as funding for global research on the disease, sent a petition to the FDA asking for early approval of Relyvrio/Albrioza. The petition, signed by more than 50,000 people affected by ALS, urged the U.S. regulator not to delay the new drug because the community cannot wait another few years for one more clinical trial, patients would simply die.
In late May 2022, a letter signed by four dozen physicians was sent to the FDA requesting approval of Relyvrio/Albrioza. According to physicians, the regulator may delay approval of the drug to wait for the results of the phase 3 clinical trial to be ready. If the study confirms the efficacy of the treatment, many ALS patients will actually be deprived of the possibility of extending their lives. If the regulator approves Relyvrio/Albrioza without any delay and the clinical trial results do not demonstrate its benefit, patients will be treated with a possibly ineffective drug, but at least one that is safe and does not cause significant harm.
Canada
In mid-June 2021, Amylyx submitted a New Drug Submission (NDS) to Health Canada to approve Albrioza against amyotrophic lateral sclerosis.
In mid-June 2022, Albriosa received approval from Health Canada to treat amyotrophic lateral sclerosis in adult patients.
Albrioza has obtained marketing approval conditionally, meaning the drug has yet to definitively validate its own efficacy and safety.
Because there are no clinical data on the use of Albrioza in the pediatric population, Albrioza is not indicated for the treatment of patients under 18 years of age. Clinical data on the use of Albrioza in patients 65 years of age and older are limited.
Albrioza is contraindicated in patients with hypersensitivity to the drug components, including bile acids, as well as in pregnant and breastfeeding women.
During the first 3 weeks of treatment, Albrioza is taken once a day for 1 sachet, its contents diluted in a glass of water, then the dose is increased to 1 sachet twice a day.
- Albrioza (sodium phenylbutyrate + ursodoxicoltaurine). Product monograph including patient medication information/Prescribing information. Canada. [PDF]
The Canadian price of Albrioza is set at C$18,403 ($13,340) for a one-month course of treatment. The one-year therapy costs C$224,000 ($162,400).
Albrioza will be on sale by August 2022.
- On July 29, 2022, Amylyx announced the commercial availability of Albrioza in Canada. The drug went on sale at full price.
Those hoping for free Albrioza from the Canadian public health insurance system will have to wait much longer because of the country’s complicated prescription drug reimbursement system that requires price negotiation with each of the country’s 13 states, a process that typically takes 6 to 9 months.
Europe
In early January 2022, Amylyx has submitted a Marketing Authorization Application (MAA) for Relyvrio/Albrioza to the European Medicines Agency (EMA).
Relyvrio/Albrioza Price
In the U.S., the cost of Relyvrio for uninsured patients is $12,500 for a 28-day course of treatment, or approximately $158,000 per year. Amylyx is committed to providing financial assistance to bring the co-pay down to zero for people with commercial insurance. Uninsured or underinsured patients who meet certain criteria will be able to receive Relyvrio for free.
In Canada, Amylyx has set the price of Albrioza at C$18,400 ($13,340) per month. A year’s treatment would cost C$224,000 ($162,400).
Obviously, the cost of treating ALS with Relyvrio/Albrioza should have been comparable to the cost of the intravenous Radicava (edaravone) or oral Radicava ORS (edaravone) by Japan’s Mitsubishi Tanabe Pharma.
A year’s treatment of ALS with Radicava or Radicava ORS, which doubles survival, costs about $175,000 in the United States and C$120,000 ($87,000) in Canada.
In Europe, Radicava or Radicava ORS are not approved: In late May 2019, Mitsubishi Tanabe withdrew the application for a marketing authorisation for Radicava after the European Medicines Agency (EMA) required an additional 12-month clinical trial to confirm its efficacy.
The U.S. Institute for Clinical and Economic Review (ICER) estimates that the fair cost of treating amyotrophic lateral sclerosis with Relyvrio should not exceed $170,000 per year. The experts noted that “policymakers should debate short-term pricing options, including a far lower price close to the cost of production, until the benefits of treatment can be adequately evaluated.”
- According to revised estimates of the fair price of Relyvrio, it should fall within the range of $9,100 to $30,600 per year.
Obviously, in a free market, no one has the right to tell Amylyx what price to charge for the ALS drug that is so necessary to mankind. It’s a business, and health is nothing more than an application point and a feature of commercial practices of any pharmaceutical company.
Amylyx Pharmaceuticals’s Business
The private financial injections into Amylyx has already exceeded $200 million.
In mid-December 2021, Amylyx filed for an initial public offering (IPO) on the NASDAQ stock exchange, planning to raise $100 million in investment. The money will be used to address regulatory issues and the pre-commercial launch of Relyvrio/Albrioza, as well as to carry out its clinical trial in the treatment of Alzheimer’s disease. At the beginning of January 2022, Amylyx became a public company (AMLX): at the end of February, the market value of the company was $1.7 billion.
Interest in amyotrophic lateral sclerosis treatment will only increase as a bill that provides $500 million over 5 years from the U.S. federal budget to support research by those drug manufacturers developing drugs to prevent and treat ALS is nearing approval.
- In late December 2021, the “Accelerating Access to Critical Therapies for ALS Act” (H.R. 3537) was signed into law by the American president.
The commercial outlook for Relyvrio/Albrioza and the financial future of Amylyx is very optimistic, given that there are at least 200,000 people with amyotrophic lateral sclerosis worldwide, 25,000 of whom live in the United States, 30,000 in Europe, and 3000 in Canada.
Patent protection for Relyvrio/Albrioza extends through 2033 (composition of matter) and 2040 (for the ALS treatment based on clinical trial data). Relyvrio/Albrioza is protected against generic copies in Canada (for 8 years), the U.S. (7 years), and Europe (10 years).
According to industry forecasts, Relyvrio/Albrioza will quickly become a blockbuster; the drug could reach $1.6 billion in annual sales by 2026.
Amylyx is also going to study the applicability of Relyvrio/Albrioza in the treatment of Wolfram syndrome (an inherited condition combining diabetes insipidus, childhood-onset diabetes mellitus, optic atrophy, and deafness; DIDMOAD) and for another as yet undisclosed indication.
Relyvrio/Albrioza: Efficacy and Safety of Amyotrophic Lateral Sclerosis Treatment
The CENTAUR (NCT03127514) phase 2/3 (randomized, double-blind, placebo-controlled, multicenter) clinical trial enrolled adult patients (n=137) with amyotrophic lateral sclerosis (sporadic or familial) whose manifestation of symptoms occurred within 18 months.
Among the main inclusion criteria: manifestation of ALS symptoms ≤ 18 months before the study and damage of upper and lower motor neurons in at least three body regions, indicating rapidly progressing disease; slow vital capacity (SVC) > 60% of the predicted value for gender, height, and age. Patients could continue to take riluzole and/or edaravone.
Participants were given twice-daily Relyvrio/Albrioza or a placebo for 24 weeks.
The primary endpoint was the rate of decline in the total score on the Amyotrophic Lateral Sclerosis Functionality Rating Scale–Revised (ALSFRS-R), which assesses disease progression and severity.
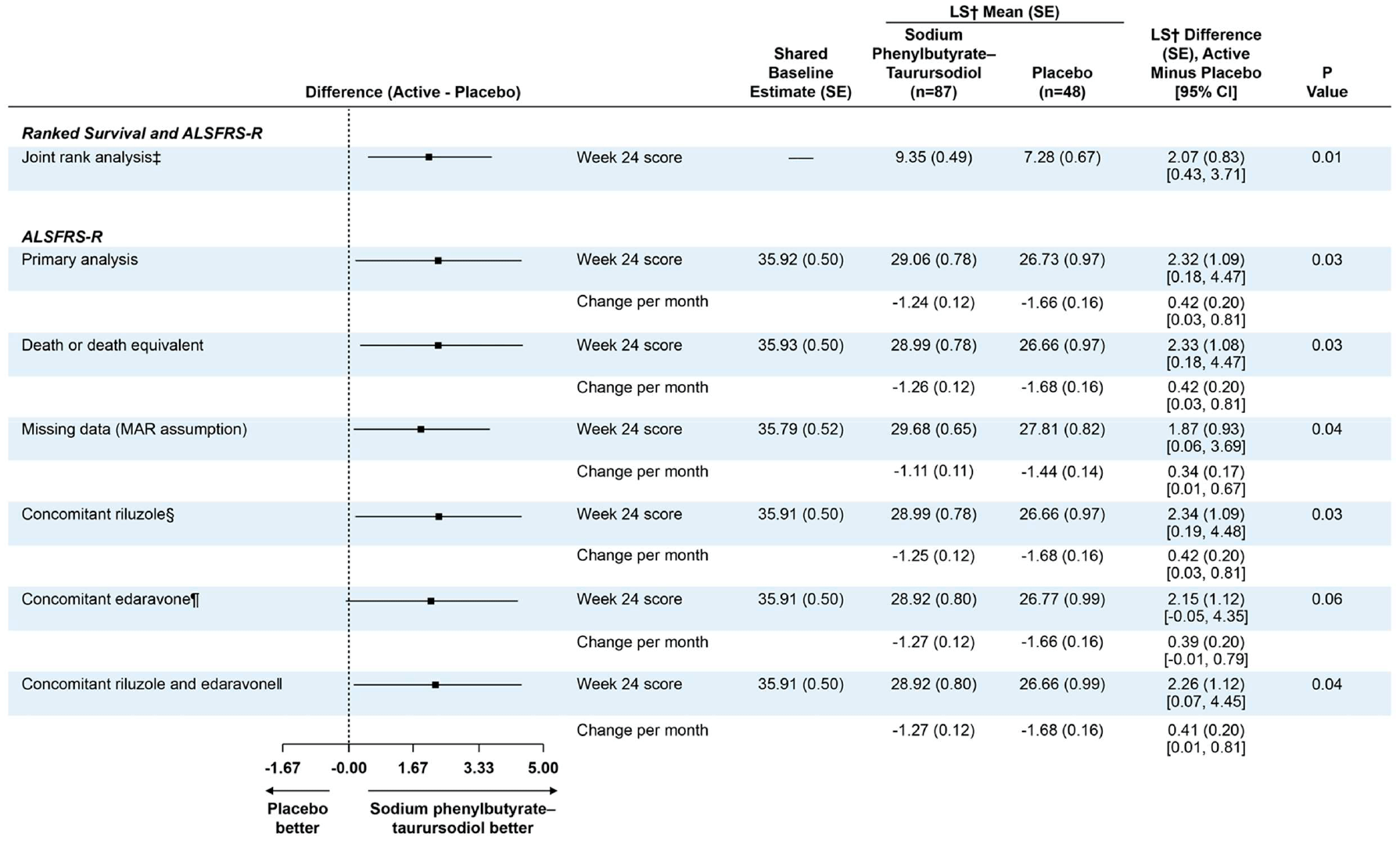
The least-square mean rate of change in the ALSFRS-R score per month was −1.24±0.12 points in the Relyvrio/Albrioza group — versus −1.66±0.16 points in the control group. The difference was 0.42 (95% CI: 0.03 to 0.81) per month, which was both a statistically significant (p=0.03) and an indication of a beneficial effect of Relyvrio/Albrioza.
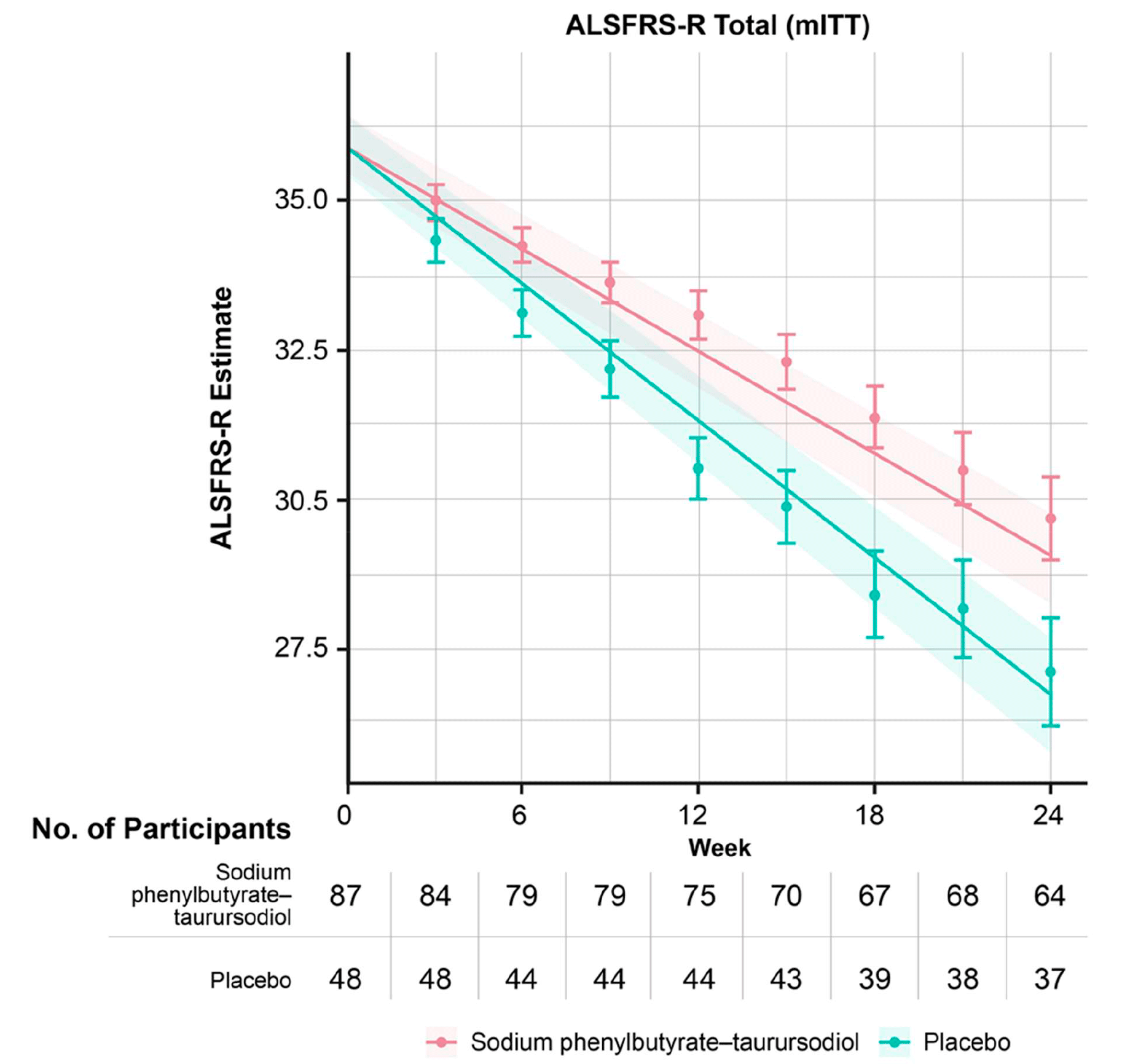
Regarding the final change in ALSFRS-R score relative to baseline at the end of the study period, the difference of 2.32 points (95% CI: 0.18 to 4.47) was statistically significant (p=0.03).
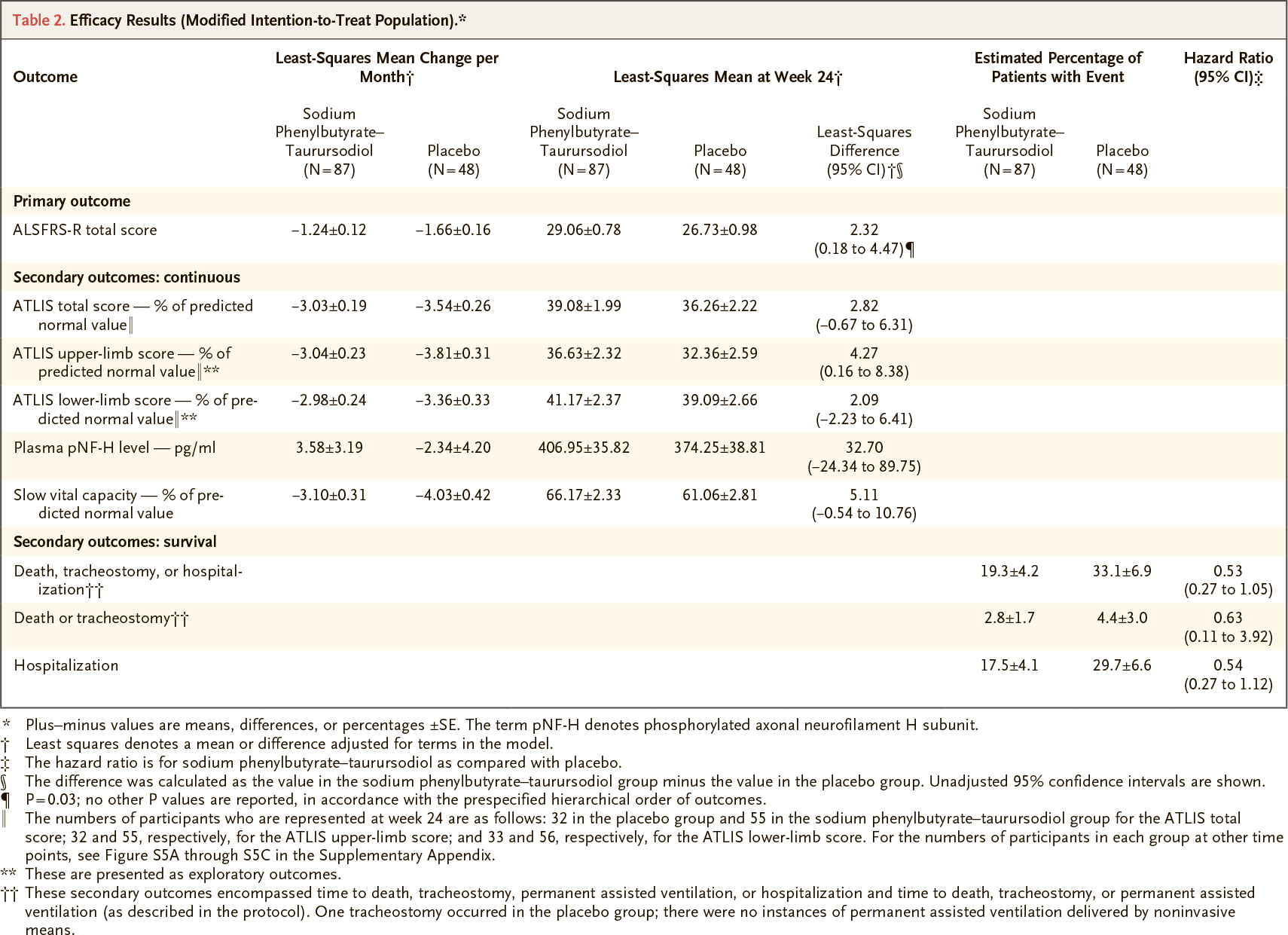
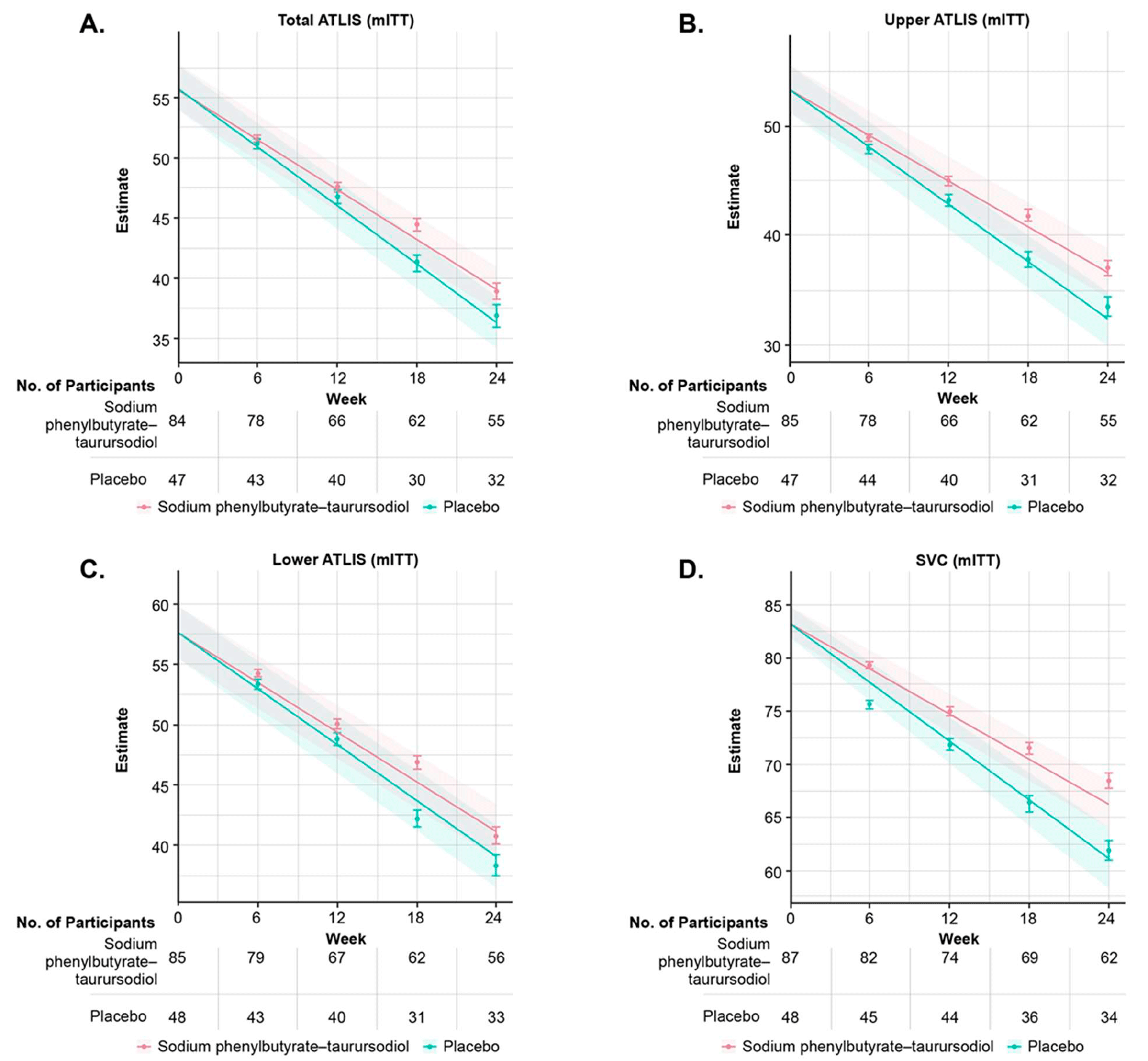
Other measures of Relyvrio/Albrioza efficacy that outperformed those in the control group (numerically, but not statistically) included the following:
- Reduced rate of decline in aggregate muscle strength: the mean change in total score on the Accurate Isometric Limb Muscle Strength Test (ATLIS) scale per month was −3.03% — versus −3.54% (p=0.12)
- Delayed progression of pulmonary function deterioration: the mean decrease in SVC score per month was 3.10% of the predicted normal value — vs. 4.03% (p=0.08)
- Improved survival: reduced cumulative odds of death, tracheostomy, or hospitalization: hazard ratio (HR) 0.53 (95% CI: 0.27 to 1.05; p=0.09).
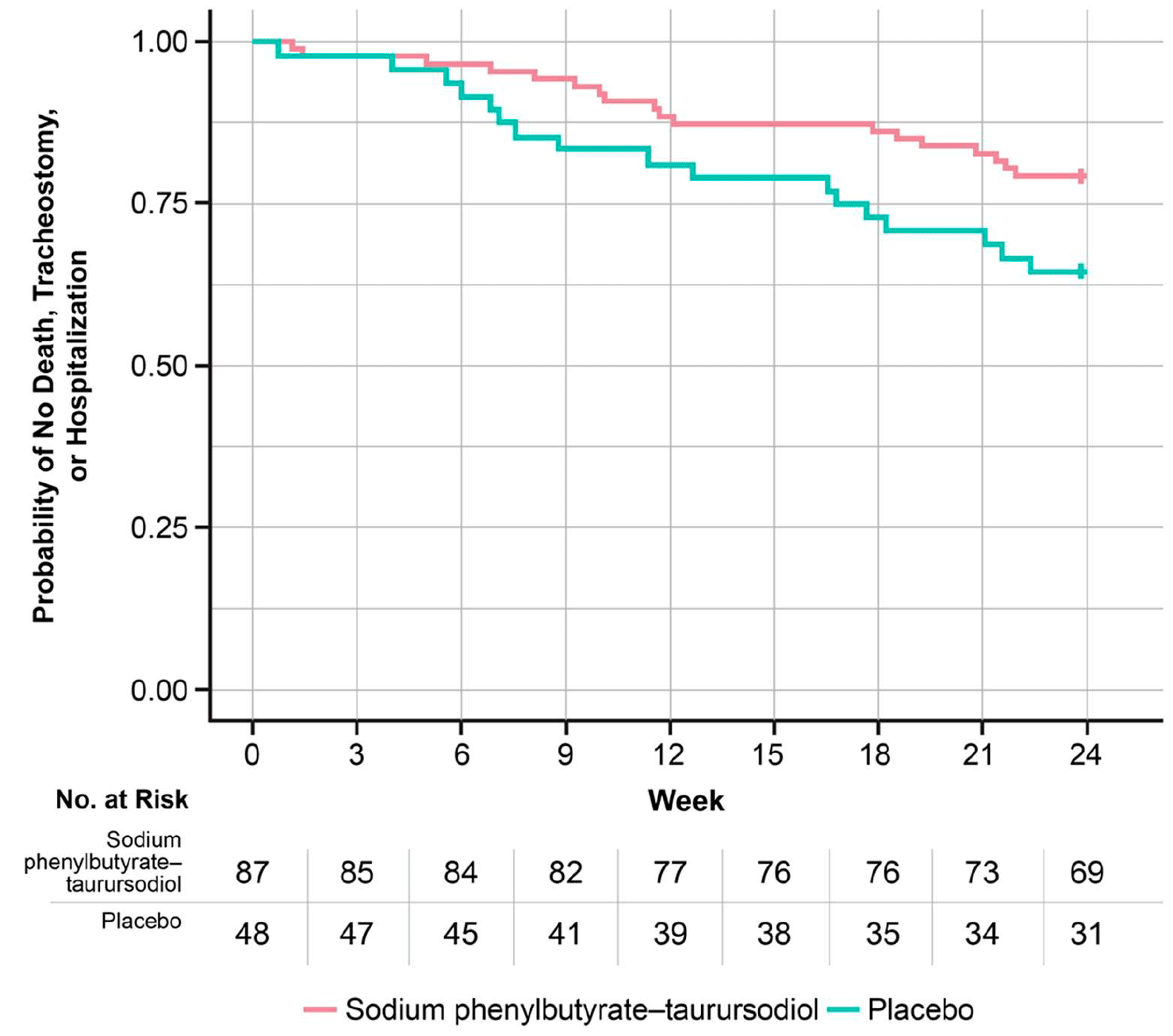
Patients who completed the study could continue to participate in open-label extension study CENTAUR-OLE (NCT03488524) when all received Relyvrio/Albrioza. At follow-up, at a maximum of 35 months from randomization, median overall survival (OS) came out to 25.0 months (95% CI: 19.0 to 33.6) in the group that initially received Relyvrio/Albrioza and 18.5 months (95% CI: 13.5 to 23.2) in the group that initially received placebo (HR 0.56 [95% CI: 0.34 to 0.92]; p=0.023). Thus, Relyvrio/Albrioza, which reduced the risk of death by 44%, provided a median prolongation of overall survival of 6.5 months.
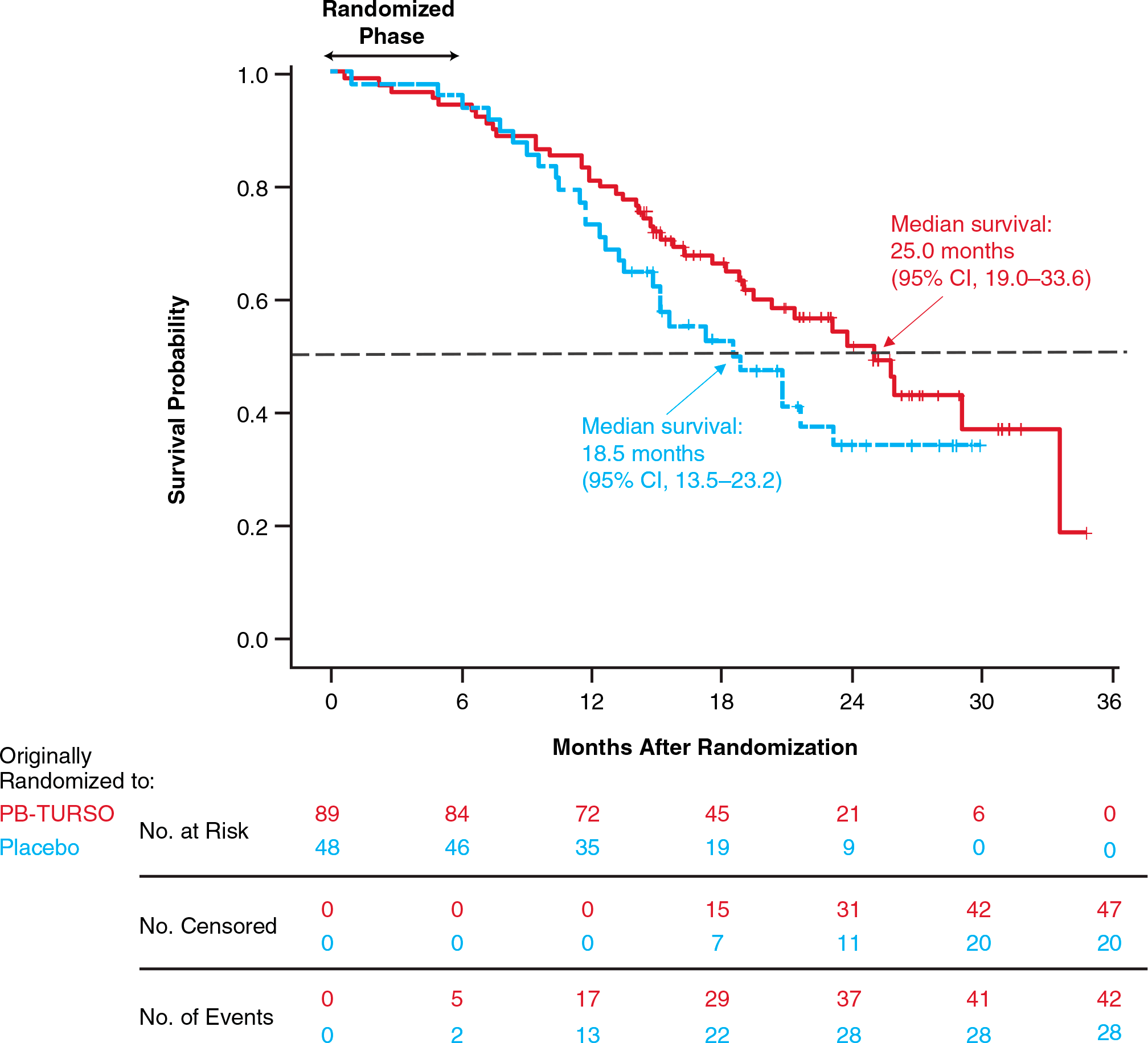
The estimated probability of 12-month survival was 80.9% (95% CI: 71.1 to 87.7) and 72.9% (95% CI: 58.0 to 83.3) in the Relyvrio/Albrioza and placebo groups, respectively; 24-month survival was 51.6% (95% CI: 38.9 to 62.9) and 33.9% (95% CI: 19.4 to 49.1).
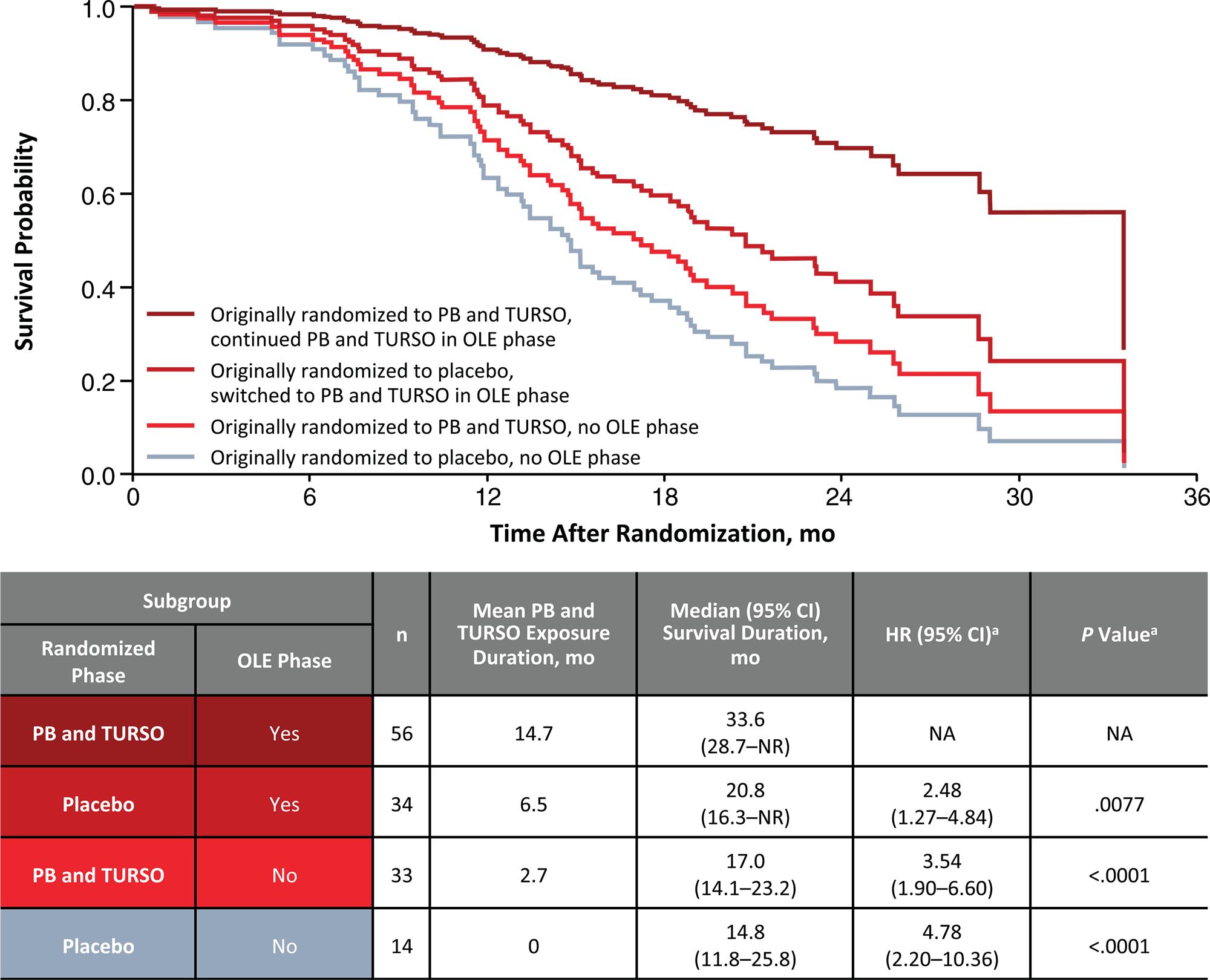
An additional analysis found that the duration of use of Relyvrio/Albrioza is more important for prolonging survival in patients with amyotrophic lateral sclerosis than the current severity of the disease. It is possible that this drug, which restrains motor neuron death, will be effective in all stages of ALS.
Thus, according to a post hoc analysis of CENTAUR and CENTAUR-OLE data, for patients (n=56) who received Relyvrio/Albrioza throughout the study (mean 14.7 months), median survival was 33.6 months (95% CI: 28.7 to NR), whereas for patients (n=14) who received placebo, it came to 14.8 months (95% CI: 11.8 to 25.8). If subjects (n=34) in CENTAUR received placebo and then in CENTAUR-OLE switched to the Relyvrio/Albrioza group, their survival was prolonged to 20.8 months (95% CI: 16.3 to NR). In other words, continuous administration of Relyvrio/Albrioza prolonged life by up to 2.3 times. If you take the drug longer, you can presumably stay alive even longer.
Relyvrio/Albrioza was characterized by acceptable tolerability, except that an increased frequency of gastrointestinal adverse events was noted in the drug group. Patients in CENTAUR experienced adverse events, such as diarrhea (18%), abdominal discomfort (10%), nausea (9%), and constipation (8%). However, these issues were mild in severity.
In the Relyvrio/Albrioza group, there was an increased frequency of adverse cardiac events and ECG abnormalities, such as atrial fibrillation, atrioventricular block first degree, bundle branch block left, left anterior hemiblock, palpitations, tachycardia, and intraventricular conduction delay. Fourteen patients (17 events) in CENTAUR and CENTAUR-OLE encountered them.
Meanwhile, the 48-week PHOENIX (NCT05021536) phase 3 (randomized, double-blind, placebo-controlled, multicenter, international) clinical trial was launched and will study Relyvrio/Albrioza in an international patient population (n=600) with amyotrophic lateral sclerosis. In contrast to CENTAUR, the inclusion criteria have been lowered: patients whose manifestation of symptoms occurred within 24 months are eligible to participate. The study should be completed by mid- to late-2024.
Relyvrio/Albrioza: Mechanism of Action
Relyvrio/Albrioza, known by the code name AMX0035, is a proprietary oral combination of two drugs long used for medical indications, sodium phenylbutyrate (PB) and tauroursodeoxycholic acid (TUDCA).
Sodium phenylbutyrate is used for treating several diseases associated with disorders of the urea cycle. As a prodrug, phenylbutyrate is rapidly metabolized to phenylacetate, which, by conjugating with glutamine, forms phenylacetylglutamine. The latter is excreted by the kidneys. Since on a molar level phenylacetylglutamine is comparable to urea (both contain two moles of nitrogen), the former provides alternative transport to the latter for nitrogen excretion.
- Sodium phenylbutyrate is sold under brand names such as Buphenyl, Pheburane, Ammonaps, and triButyrate.
Tauroursodeoxycholic acid, also known as taurursodiol, ursodoxicoltaurine, and ursodeoxycholyltaurine, is a naturally-occurring bile acid that is formed endogenously in the liver by conjugating taurine with ursodeoxycholic acid (UDCA). For centuries, it has been used in Oriental medicine to dissolve gallstones and for treating various liver diseases.
- Taurursodiol is marketed in some countries under the brand names Taurolite, Tauro, Tudcabil, and Taursol.
Numerous molecular mechanisms are believed to contribute to the pathogenesis of neurodegenerative diseases such as amyotrophic lateral sclerosis or Alzheimer’s disease, which have a harmful effect on neurons. A key role here is played by the biochemical dysfunction of the endoplasmic reticulum (ER) and mitochondria, which are physically and functionally linked through mitochondrial-associated membranes (MAM) involved in Ca2+ signaling, lipid transport, energy metabolism, and cell survival. In addition to their crucial physical connection coordinating the signaling pathways of the cell cycle of life and death, any loss of contact between them complicates and worsens the prevailing conditions in any neurodegenerative disorder. [1] [2]
The presumed mechanism of action of Relyvrio/Albrioza is as follows. Because sodium phenylbutyrate works as a low-molecular-weight chaperone, it suppresses toxic responses to ER stress and inhibits neuronal death induced by accumulating misfolded or mutant proteins. It is also a histone deacetylase (HDAC) inhibitor that, through epigenetic regulation, enhances the expression of heat shock proteins. [1] [2] [3]
Tauroursodeoxycholic acid restores mitochondrial bioenergetic deficits through numerous processes, including by inhibiting the translocation of the BAX protein into the mitochondrial membrane. It, by actually reducing the permeability of the mitochondrial membrane, inhibits the release of cytochrome c and other pro-apoptotic factors leading to the activation of caspases. The latter induces cell death. In other words, tauroursodeoxycholic acid ensures the growth of the apoptotic threshold of the cell. [4] [5]
According to preclinical data, sodium phenylbutyrate and tauroursodeoxycholic acid show activity in mouse models of neurodegeneration.
Thus, sodium phenylbutyrate reduced the number and size of amyloid plaques in the cortex and hippocampus of APPSwe/PSEN1dE9 transgenic mice (serve as Alzheimer’s disease models), which led to improved cognitive performance in a spatial memory task (maze passing). [6] In a mouse model of amyotrophic lateral sclerosis, sodium phenylbutyrate, which inhibits motor neuronal death and progression of the corresponding pathology, prolonged survival when used alone and in combination with riluzole. [7] [8] In mouse models of multiple systemic atrophy and Parkinson’s disease overexpressing toxic alpha-synuclein, sodium phenylbutyrate protected against neurodegeneration. [9] [10]
Tauroursodeoxycholic acid reduced amyloid beta deposits in the cortex and hippocampus and improved cognitive abilities (contextual fear memory) in APP/PS1 transgenic mice (serve as Alzheimer’s disease models). [11] In mouse models of Parkinson’s disease, Huntington’s disease, or amyotrophic lateral sclerosis, tauroursodeoxycholic acid improved pathological patterns and neurobehavior. [12]
When it comes to the combination of sodium phenylbutyrate and tauroursodeoxycholic acid studied on primary skin fibroblasts from patients with sporadic ALS, the combination altered many more genes and metabolites than each compound alone. Most of the changes were unique to the combination and affected the expression of genes involved in nucleocytoplasmic transport (NCT), unfolded protein response (UPR), mitochondrial processes, innate immunity functions, nucleic acid metabolism, and RNA processing. The drug combination resulted in the following effects: [13]
- downregulation of S-adenosylmethionine levels: as a methyl donor for histone and DNA methyltransferases, it affects epigenetics
- downregulation of significantly altered nucleoporin genes: abnormal nuclear membrane shape, loss or mislocalization of nuclear pore complex protein, dysregulation of NCT dynamics were observed in ALS models
- upregulation of genes promoting survival in ER stress conditions
- downregulation of mediators of UPR signaling
- upregulation of several subunits of mitochondrial respiratory chain
- changes in the expression of genes involved in the activation of the innate immune system: STING1, cGAS, 20S and 26S subunits of immunoproteasome
- downregulation of genes related to RNA polymerase II (RNAP II) transcription: for example, one of these genes, SETX, encodes senataxin, which modulates RNAP II binding to chromatin and which is associated with juvenile ALS.
In other words, the combined effect of sodium phenylbutyrate and tauroursodeoxycholic acid, beneficial in ALS, is characterized by an advantage over the effect of a single molecule.
The Relyvrio/Albrioza formulation contains 3 g of sodium phenylbutyrate and 1 g of tauroursodeoxycholic acid.
The idea behind Relyvrio/Albrioza came from Joshua Cohen and Justin Klee, the founders of Amylyx, back in 2013 when they were students at Brown University.
Relyvrio/Albrioza: Expert Comments
Based on the ALSFRS-R clinical changes, Relyvrio/Albrioza was associated with an approximately 25% reduction in the rate of worsening of the natural course of ALS. Yes, the claimed therapeutic effect is very modest, but given the critical demand for new drugs, it should be taken with proper optimism.
It should be understood that a decrease of even 1–2 points on the ALSFRS-R scale can be reflected in a significant deterioration in patients’ ability to act independently. Because the ALSFRS-R assesses all types of everyday functions (including the ability to walk, dress, eat, talk, and breathe), such impairments manifest themselves in different ways. For example, someone may lose the ability to move around unaided or even go to bed, or someone may lose the ability to eat as usual, having to switch to feeding through a tube.
A 44% reduction in the risk of death is an excellent result: Considering that the median survival rate for ALS is approximately 4 years from the manifestation of symptoms, this means that life with Relyvrio/Albrioza can be prolonged up to 6–6.5 years.
The therapeutic effect of Relyvrio/Albrioza has been confirmed to be independent of the background use of riluzole and/or edaravone, which are approved for ALS. They were taken by 77% of the study participants. A sensitivity analysis adjusted for concomitant use of riluzole showed an estimated difference between the Relyvrio/Albrioza group and the placebo group of 2.34 points (95% CI: 0.19 to 4.48; p=0.03), edaravone of 2.15 points (95% CI: −0.05 to 4.35; p=0.06), both drugs of 2.26 points (95% CI: 0.07 to 4.45; p=0.04).
The failures of many other clinical trials of experimental treatments for amyotrophic lateral sclerosis are difficult to explain due to the lack of informative biomarker data. This study evaluated the extent of changes in the plasma concentration of phosphorylated axonal neurofilament heavy chain (pNF-H): the increase in this protein is thought to correlate with motor neuron degeneration. The Relyvrio/Albrioza group was found to have a mean increase in plasma pNF-H concentration of 3.58 pg/ml per month, whereas, in contrast, the placebo group had a 2.34 pg/ml drop per month. In other words, there was a discrepancy between a positive clinical effect and no effect on the biomarker, indicating a potential pharmacodynamic effect in ALS. It is unclear whether this is due to the peculiarities of the performed statistical analysis of the data or the truly limited effect of Relyvrio/Albrioza on the ongoing neurodegenerative disease process.
It remains unknown whether the therapeutic effect shown by Relyvrio/Albrioza is due to the action of its individual components (sodium phenylbutyrate or taurursodiol), their combination, or related to specific doses. The ongoing NCT03800524 phase 3 clinical trial, which is testing only taurursodiol and is expected to be completed in late 2022, may provide some answers.
Considering some questions about the efficacy of Relyvrio/Albrioza and the ability of patients to continue taking it, additional, longer, and larger clinical trials are needed. In doing so, they must strike a balance between the scientific rigor of the study and the generalizability of the findings to show the drug’s suitability in a broad population of patients with ALS. Of course, since Relyvrio/Albrioza has entered the commercial market, such confirmatory clinical trials may be postponed indefinitely, especially since the drug components have long been available as stand-alone drugs.
Extras
Prescribing Information
Relyvrio (sodium phenylbutyrate + taurursodiol). Prescribing information. U.S. [PDF]
Albrioza (sodium phenylbutyrate + ursodoxicoltaurine). Product monograph including patient medication information/Prescribing information. Canada. [PDF]
Scientific Data
Survival analyses from the CENTAUR trial in amyotrophic lateral sclerosis: Evaluating the impact of treatment crossover on outcomes. Muscle Nerve. 2022 May 4. [source]
Long-term survival of participants in the CENTAUR trial of sodium phenylbutyrate-taurursodiol in amyotrophic lateral sclerosis. Muscle Nerve. 2021 Jan;63(1):31-39. [source]
Trial of sodium phenylbutyrate-taurursodiol for amyotrophic lateral sclerosis. N Engl J Med. 2020 Sep 3;383(10):919-930. [source]
Effects of PB-TURSO on the transcriptional and metabolic landscape of sporadic ALS fibroblasts. Ann Clin Transl Neurol. 2022 Sep 9. [source]
FDA Advisory Committee
AMX0035. Combined FDA and applicant briefing document. Peripheral and central nervous system (PCNS) Drugs Advisory Committee (DAC) meeting, March 30, 2022. [PDF]
AMX0035. Clinical overview. Peripheral and central nervous system (PCNS) Drugs Advisory Committee (DAC) meeting, March 30, 2022. [PDF]
Investor Relations
Amylyx Pharmaceuticals. Relyvrio. FDA approval conference call. September 30, 2022. [PDF]
Amylyx Pharmaceuticals. Company overview. September 2022. [PDF]
Amylyx Pharmaceuticals. Company overview. June 2022. [PDF]
Amylyx Pharmaceuticals. Company overview. April 2022. [PDF]
Amylyx Pharmaceuticals. Company overview. February 2022. [PDF]