Highlights
Zynteglo (betibeglogene autotemcel) is a gene therapy to treat transfusion-dependent beta-thalassemia.
Zynteglo made its debut in early June 2019, when the European Medicines Agency (EMA) granted conditional approval to Bluebird Bio for gene therapy of transfusion-dependent beta-thalassemia in patients 12 years old and older without the β0/β0 genotype who are eligible for a hematopoietic stem cell (HSC) transplantation but have no HLA-matched HSC donor.
In late March 2022, Zynteglo lost its authorization in Europe at the request of Bluebird, which was unhappy with the commercial failure due to the high cost of the treatment. Zynteglo, prescribed once, was priced at €1.575 million ($1.77 million). With one-fifth paid upfront in the first year, the rest was paid over the next four years and only if the treatment worked.
In mid-August 2022, Zynteglo was approved by the U.S. Food and Drug Administration (FDA), and more broadly: for the treatment of beta-thalassemia in adults and children who require regular red blood cell transfusions.
The cost of Zynteglo in the United States is set at $2.8 million. Up to 80% of the cost can be refunded if the treatment fails.

Roctavian: Gene Therapy for Hemophilia A
A single infusion of valoctocogene roxaparvovec can completely cure severe hemophilia A.
What Is Beta-thalassemia
Beta-thalassemia is a hereditary hemoglobinopathy caused by point mutations in the human beta globin (HBB) gene in which the synthesis of one or both beta globin chains is disrupted. The latter, along with the two chains of alpha globin, are part of hemoglobin A (HbA), the main variant of adult hemoglobin.
An imbalance in the ratio between alpha globin and beta globin leads to ineffective erythropoiesis and intramedullary hemolysis, when erythrocyte maturation is impaired and their developing precursors in the bone marrow are destroyed.
Clinical manifestations of beta-thalassemia include low hemoglobin levels, hemolytic anemia, impaired iron metabolism, hepatosplenomegaly, and osteoporosis.
The severity of beta-thalassemia directly depends on the zygosity and type of mutations, of which there are over two hundred, which can be both completely disabling (β0) beta globin synthesis and reducing (β+) its production. In simplified terms, a homozygous β0/β0 mutation or a heterozygous β0/β+ mutation is referred to as beta-thalassemia major (alternatively called Cooley anemia and Mediterranean anemia), when beta globin chains are not produced at all or in very insufficient amounts, which involves lifelong regular blood transfusions. Homozygous β+/β+ or heterozygous β0/β+ mutations are usually diagnosed with beta-thalassemia intermedia, heterozygous β+/β or β0/β mutations are diagnosed with beta-thalassemia minor; such forms usually do not require blood transfusions. In addition, a specific βE/β0 genotype is observed in half of transfusion-dependent beta-thalassemia cases.
Beta-thalassemia is a rare genetic disease: The prevalence of symptomatic disease is 1 case per 100,000 worldwide and 1 case per 10,000 in Europe. Approximately 60,000 children are born each year with symptomatic beta-thalassemia.
How Beta-thalassemia Is Currently Treated
In theory, beta-thalassemia can be cured by hematopoietic stem cell transplantation, but there are critically few suitable donors: they can be found for about 25%–30% of patients. Again, because of the high risk of graft rejection and graft-versus-host reactions, this procedure is usually performed only on young children.
And therefore, in most cases the disease involves chronic maintenance therapy, which includes regular (every 2–5 weeks) packed red blood cells (pRBC) transfusions (blood transfusions), iron chelating agents (to reduce high iron levels), splenectomy, induction of fetal hemoglobin.
There is also a specialized drug, Reblozyl (luspatercept), indicated for the treatment of anemia in transfusion-dependent beta-thalassemia. Luspatercept, which promotes erythrocyte maturation, reduces the burden of blood transfusions by about a third, but is associated with thrombosis. Luspatercept was developed by Acceleron Pharma, later purchased by Merck & Co.
Despite improved blood transfusion methods, the quality of life of patients with beta-thalassemia remains very low, and the increased risk of morbidity and mortality due to iron overload due to constant blood transfusions leads to an average age of death of 37 years. Iron accumulation is reflected in endocrinopathies, cardiomyopathy, and cirrhosis. The leading cause of death is heart failure due to iron accumulation in the heart.
How Zynteglo Works
The treatment with Zynteglo, known as betibeglogene autotemcel, is based on a single intravenous infusion of a gene therapy drug, after which patients with beta-thalassemia have a very high chance of permanently eliminating the need for blood transfusions.
Apheresis-extracted patient CD34+ hematopoietic stem cells are transduced ex vivo by lentiviral transfer of functional copies of the modified beta globin gene (βA-T87Q-globin). The cells are then returned to the patient who has previously undergone a chemotherapeutic myeloablative bone marrow conditioning procedure with busulfan, necessary to suppress the immune system in order to engraft the stem cells. Once engraftment has taken place and the stem cells have differentiated, the synthesis of erythrocytes containing the biologically active βA-T87Q-globin is triggered, which combines with alpha globin to produce functional hemoglobin A. Once the latter reaches normal levels, the healing effects of Zynteglo can be expected to last a lifetime.
The treatment process is not quick. It takes approximately three months to prepare the drug, and once it has been administered, the patient must stay in a hospital facility for several months, waiting for the immune system to recover.
How Effective Zynteglo Treatment for Beta-thalassemia
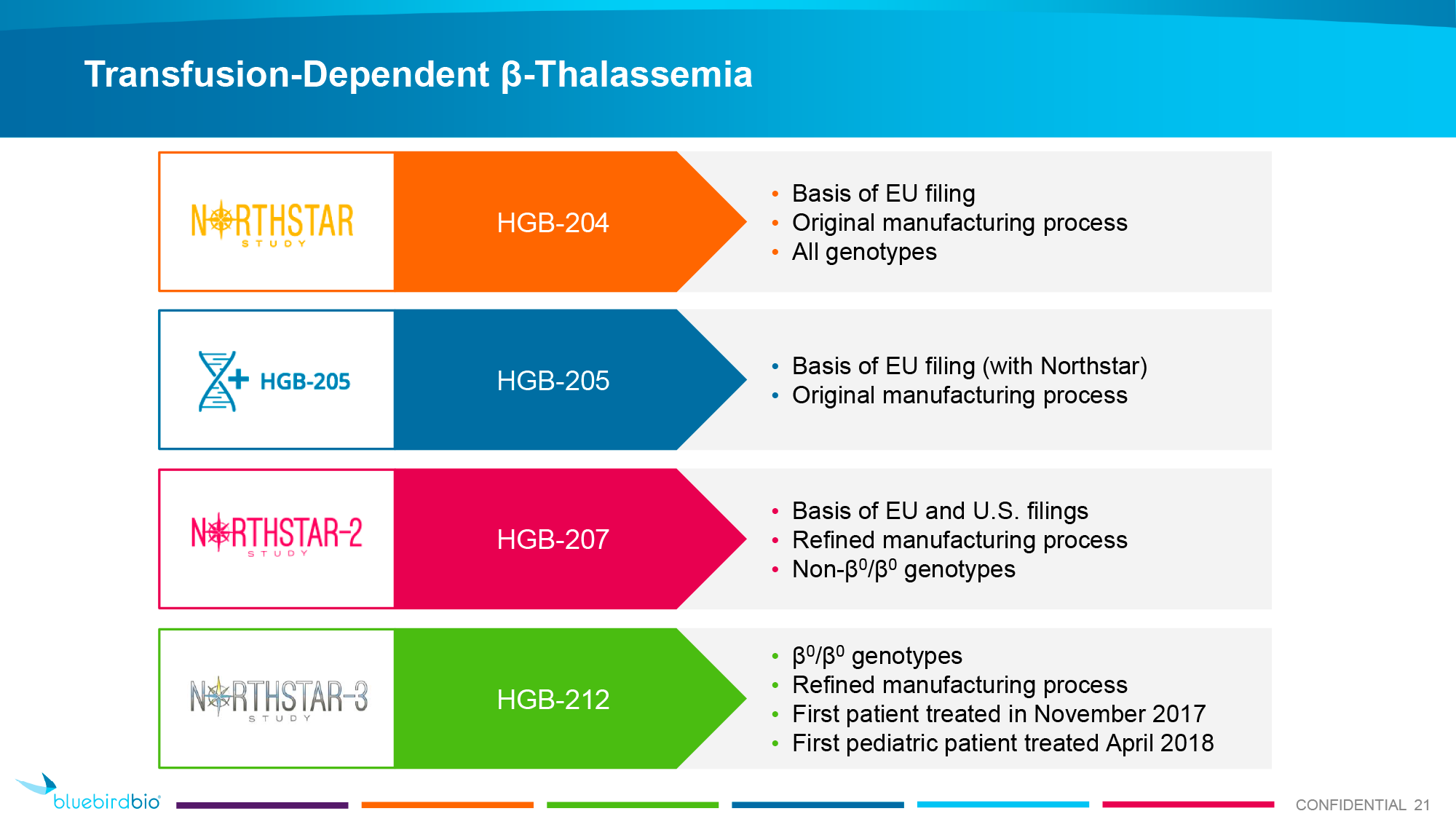
The European regulator relied on the results of multiple clinical trials of Zynteglo: completed HGB-205 (NCT02151526) and HGB-204 (Northstar, NCT01745120) phase 1/2, ongoing HGB-207 (Northstar-2, NCT02906202) and HGB-212 (Northstar-3, NCT03207009) phase 3, long-term observational LTF-303 (NCT02633943).
The primary endpoint was to achieve blood transfusion independence status: No need for transfusion procedures for a period of 12 months or more while maintaining hemoglobin levels at an average of at least 9 g/dL.
Data taken in mid-December 2018 for patients without the β0/β0 genotype showed that in HGB-205 and HGB-204, 75% of subjects (n=3/4) and 80% (n=8/10) respectively reached the primary endpoint: independence from blood transfusions lasted from 21.2+ to 56.3+ months and the weighted average hemoglobin level was 10.5 g/dL (9.3–13.2).
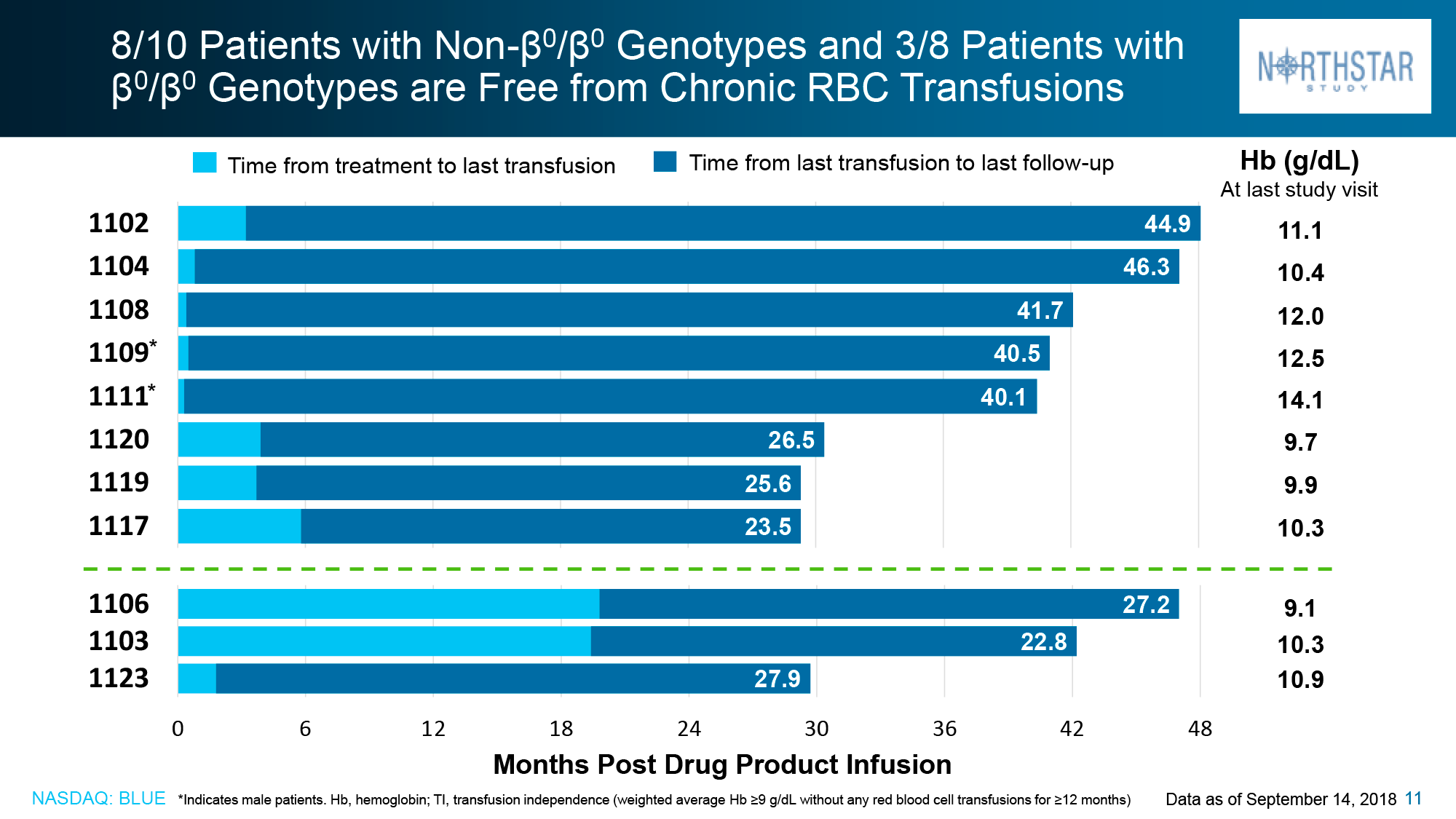
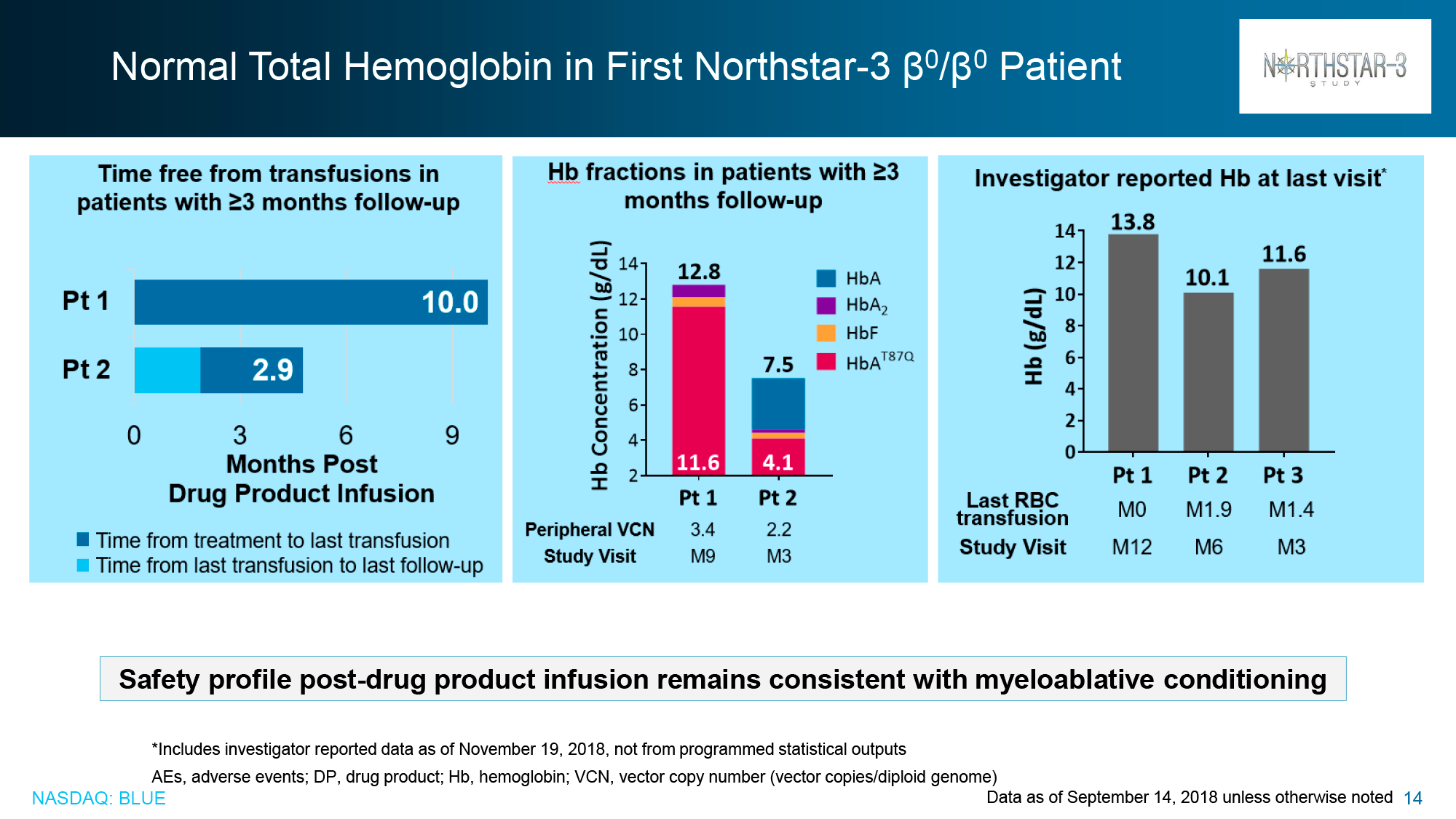
For the patients (n=3) who did not show proper therapeutic efficacy with Zynteglo, there was a 100%, 87%, and 27% reduction in blood transfusion volumes and a 100%, 85%, and 21% reduction in blood transfusion rates during the 6–24 month follow-up period.
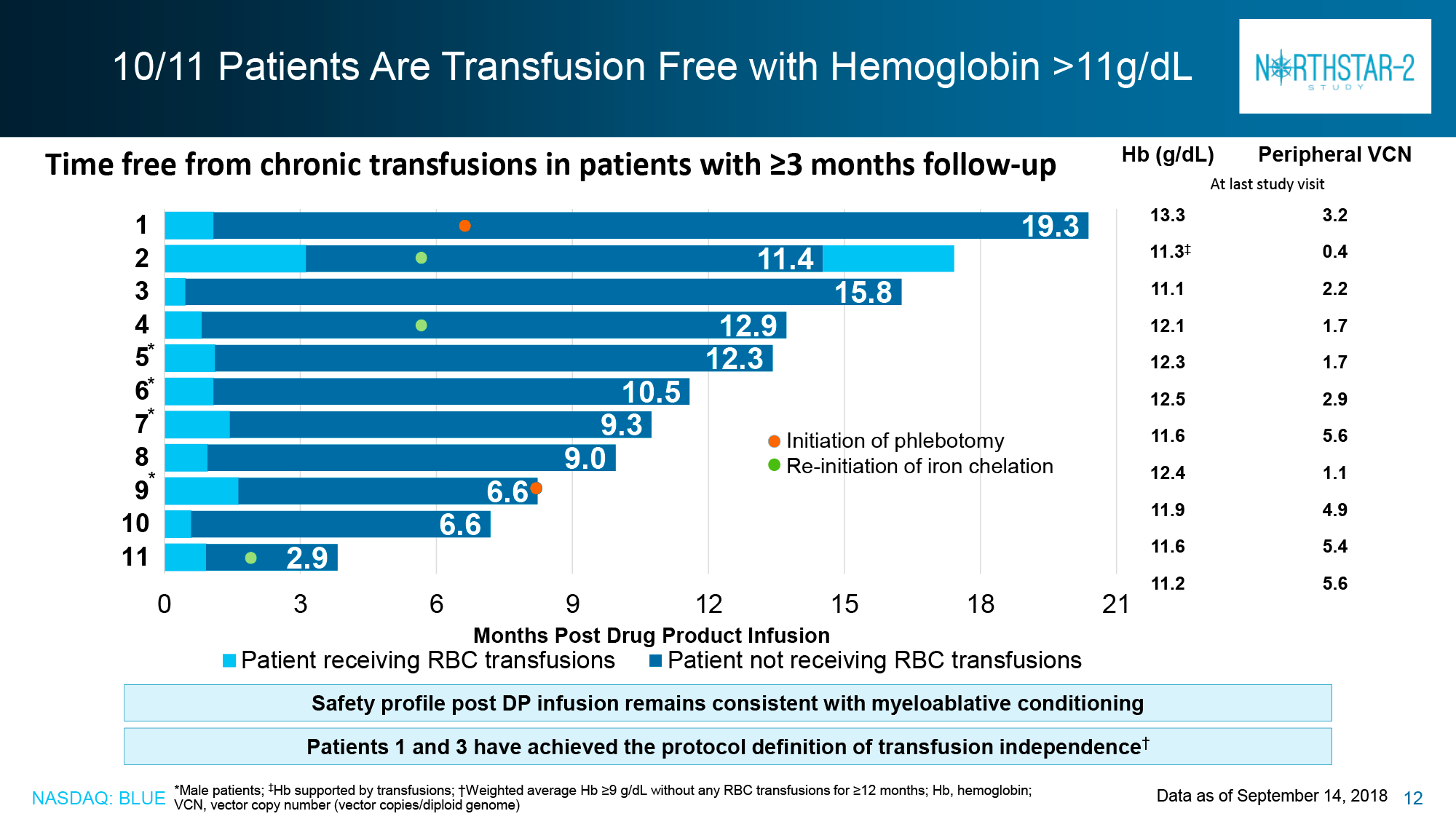
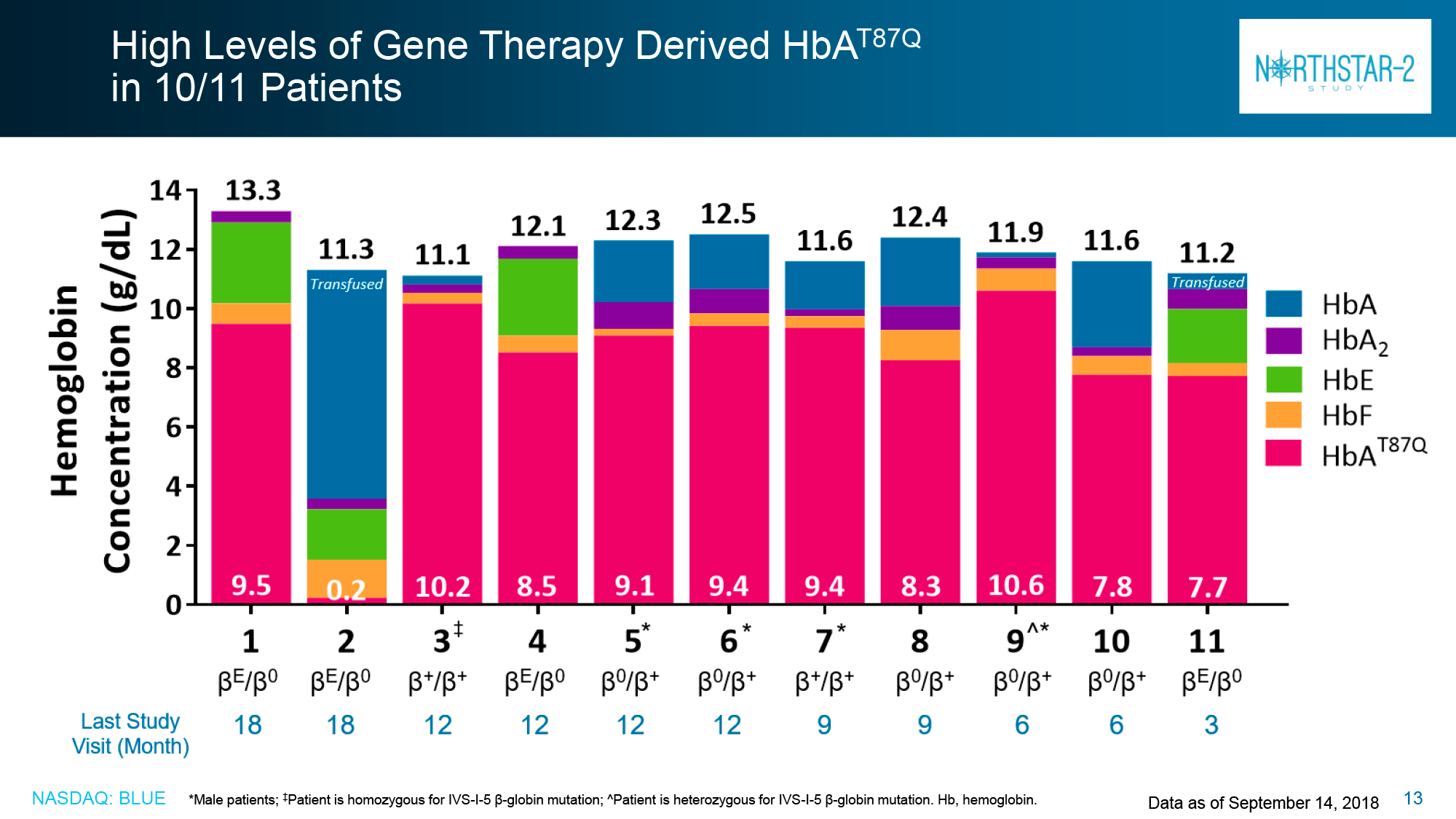
Of the HGB-207 participants without the β0/β0 genotype for whom data were collected for analysis, 80% (n=4/5) achieved claimed therapeutic status: independence from blood transfusions lasted from 12.0+ to 18.2+ months with a weighted average hemoglobin level of 12.4 g/dL (11.5–12.6).
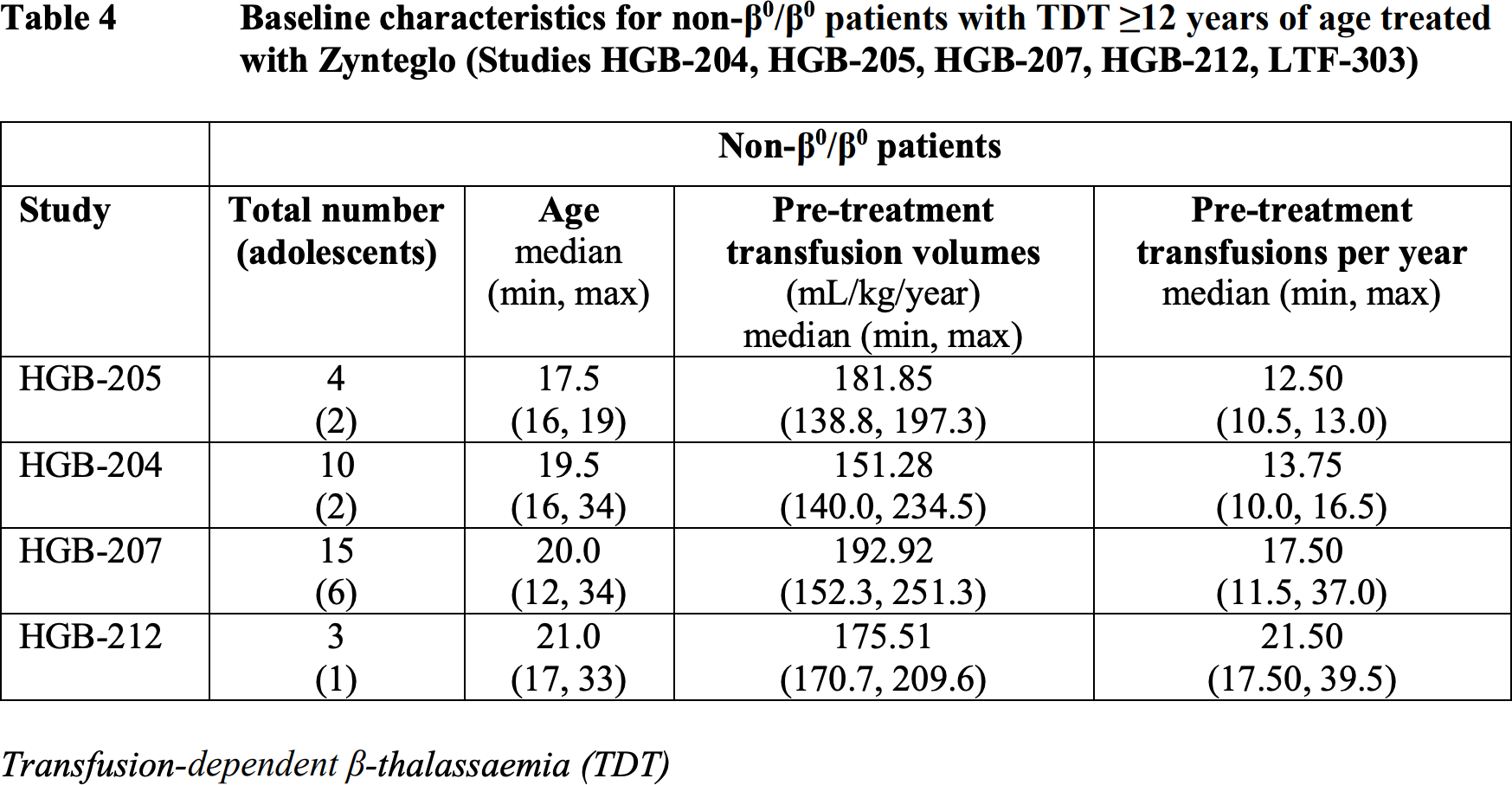
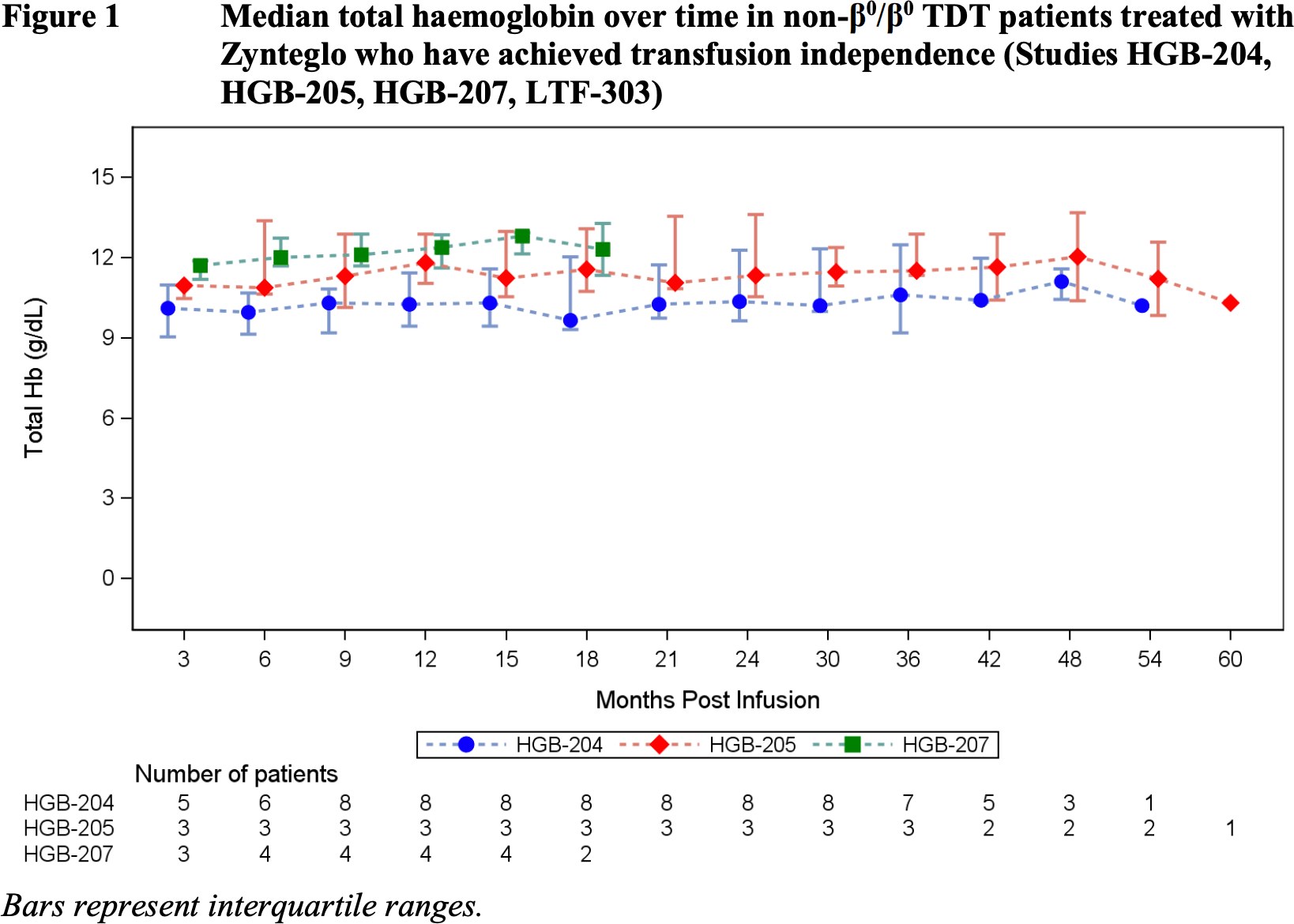
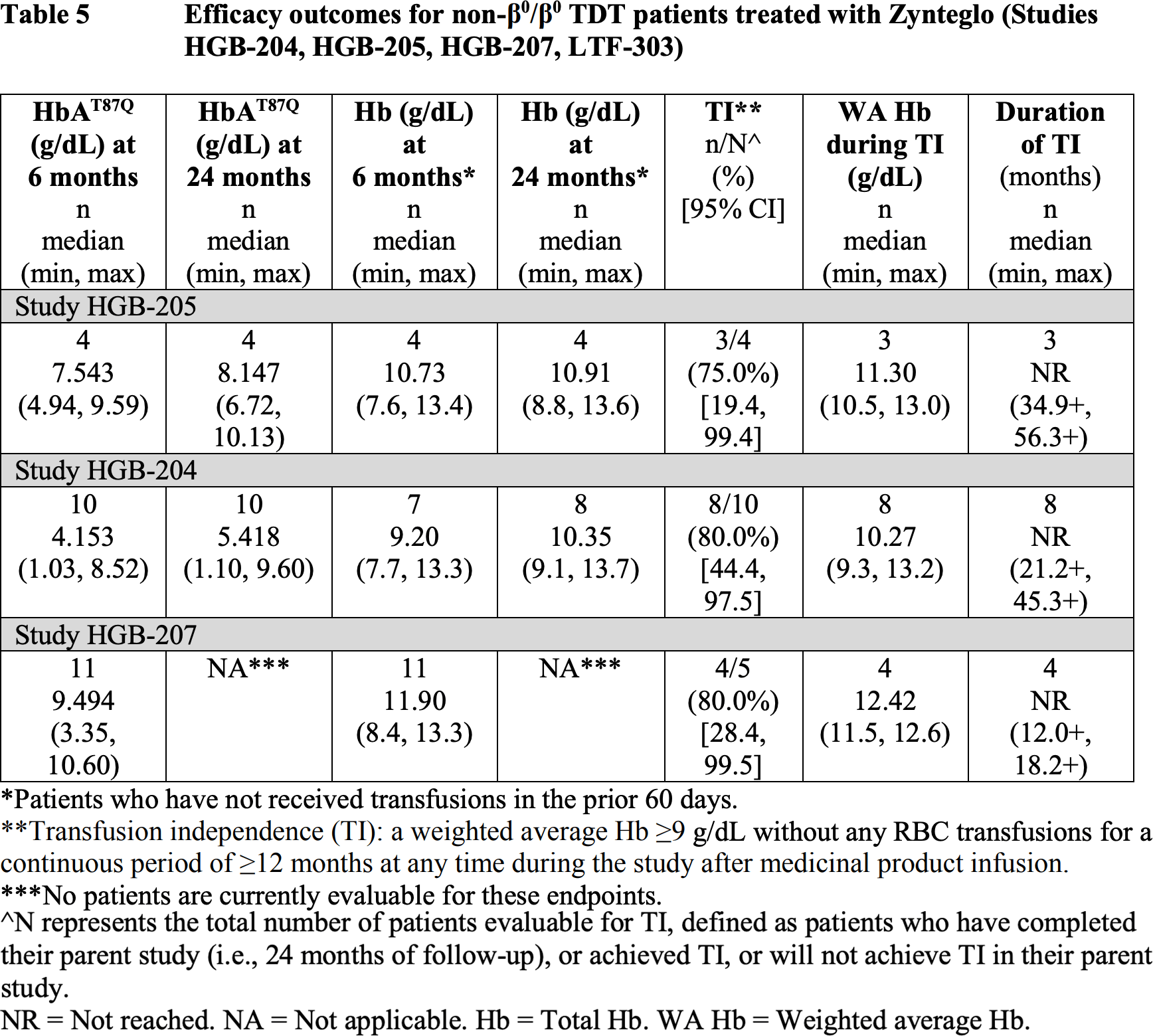
The American regulator had significantly more accumulated clinical data supporting the therapeutic efficacy of Zynteglo.
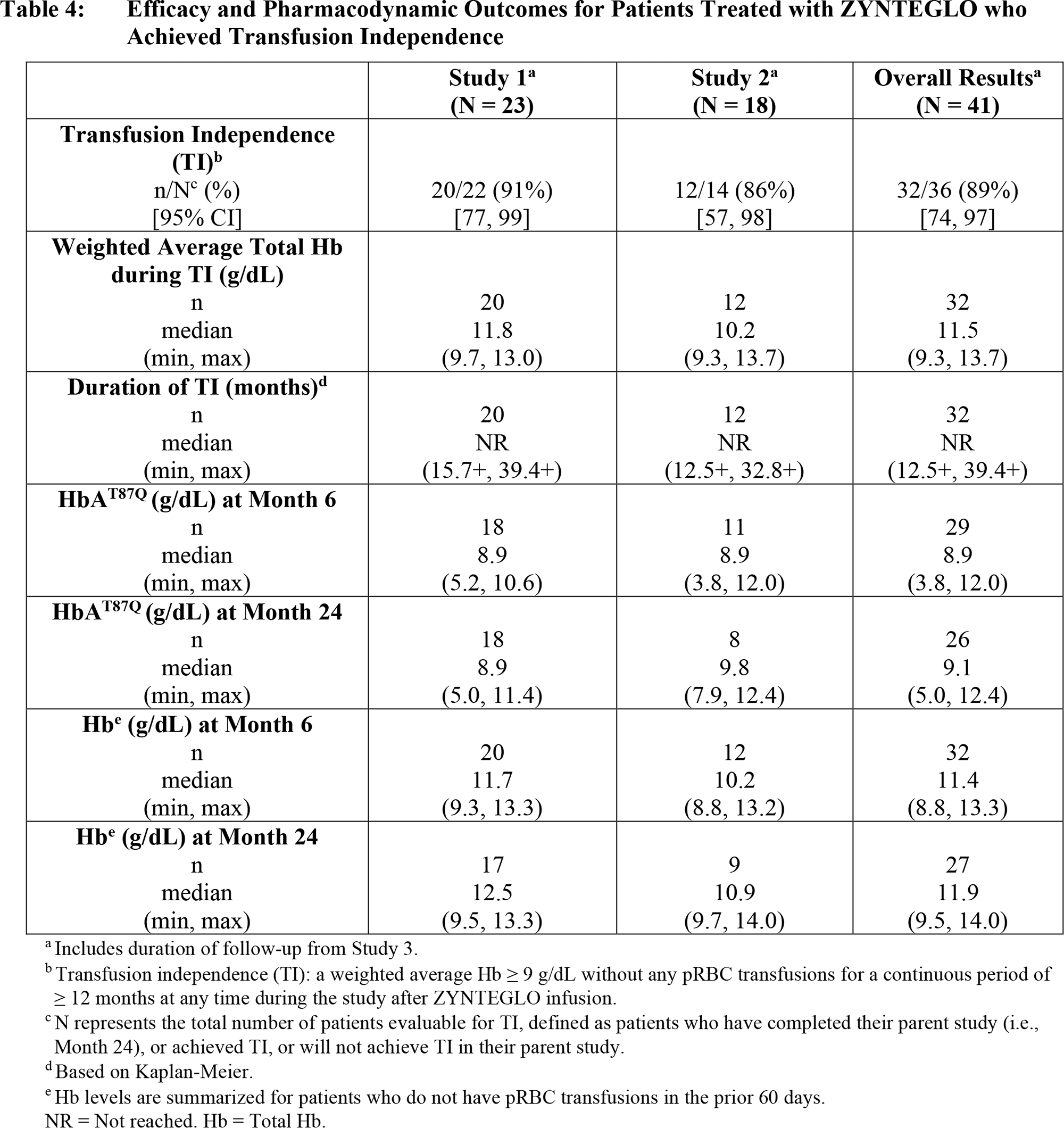
At a median follow-up of 29.5 months (13.0–48.2) and 24.6 months (4.1–35.5) for patients (n=23 and n=18) from the HGB-207 and HGB-212 clinical trials, the following results were obtained:
- primary endpoint achieved and maintained in 91% and 86% of participants, cumulatively in 89%
- median duration of independence from blood transfusions not established: 15.7+ to 39.4+ months and 12.5+ to 32.8+ months, combined 12.5+ to 39.4+ months
- weighted average hemogloblin levels were median 11.8 g/dL and 10.2 g/dL, combined 11.5 g/dL.
Patients (n=8) with the β0/β0 genotype were also included in the HGB-212 study, and 88% achieved blood transfusion independence status.
The LTF-303 long-term clinical trial is ongoing, with 13 years of follow-up for patients who agreed to participate from all other clinical trials who had been treated with Zynteglo and who had already been followed for two years. At the time of inclusion of patients (n=57) in LTF-303, transfusion independence status was fair for 81% of participants (n=46/57): 68% of patients (n=46/57) from phase 1/2 clinical trials and 89% (n=31/35) from phase 3 clinical trials.
As of the mid-August 2021 date, all 46 patients maintained the indicated status, with a median duration of 65.9 months (19.8–84.5) and 32.0 months (18.2–49.1) for patients from phase 1/2 and 3 clinical trials, respectively.
Weighted average hemoglobin levels were 10.3 g/dL (9.1–13.2) and 11.6 g/dL (9.5–13.7).
In other words, Zynteglo eliminated the need for regular blood transfusions for the vast majority of patients, while returning hemoglobin levels to normal or near-normal levels.
Prior to Zynteglo gene therapy, all patients required iron chelating agents. The LTF-303 observations showed that 59% of the subjects (n=20/34) who had achieved transfusion-independent status and still required chelation therapy after treatment, declined it at the end, and 24% (n=11/46) among those who achieved transfusion-independent status were able to undergo phlebotomy (bloodletting), another method to reduce high iron levels, but only indicated if hemoglobin levels were adequate.
Expert Comments
Bluebird understands that, first, the U.S. price of Zynteglo at $2.8 million (which, however, will be lower after procurement) was astronomical and, second, that the treatment works without a 100% guarantee of success. That’s why it promises to refund health insurance providers up to 80% of the cost of Zynteglo if the patient does not achieve and/or maintain blood transfusion independence status within two years of the drug’s infusion.
The U.S. Institute for Clinical and Economic Review (ICER) calculates that a fair price for Zynteglo is $2.1 million, with the condition that 80% of the money can be refunded if the patient becomes dependent on transfusions again within five years of treatment.
Lifetime maintenance therapy for an American patient with beta-thalassemia costs the health care system around $6.4 million.
Investment sentiment for Bluebird would be much more optimistic if Zynteglo had not left Europe, where the population of transfusion-dependent beta-thalassemia patients is 11,000 to 13,000, with half living in Italy. Commercial prospects in the United States are much more modest with their patient pool in the range of 1,300 to 1,500, with even fewer potentially eligible for gene therapy at about 850.
According to conservative industry forecasts, Zynteglo’s sales will reach $391 million in 2028.
Bluebird is finalizing a gene therapy for sickle cell disease (SCD) with lovotibeglogene autotemcel, which, like Zynteglo, relies on the exact same modified beta globin gene (βA-T87Q-globin) because the latter prevents polymerization of hemoglobin S (HbS), pathological in the disease.
Meanwhile, CRISPR Therapeutics and Vertex Pharmaceuticals are working to treat beta-thalassemia and sickle cell disease using CRISPR-Cas9 gene editing technology. The experimental exagamglogene autotemcel (CTX001) is an autologous ex vivo gene editing of patient hematopoietic stem cells to make them produce fetal hemoglobin (HbF) to compensate for low hemoglobin A (HbA) levels.
The long-term efficacy of beta-thalassemia treatment has been demonstrated: After 1.2 to 37.2 months following a single administration of exagamglogene autotemcel, 95% of patients (n=42/44) reached transfusion independence status; the remaining 5% (n=2/44) reported a 75% and 89% reduction in transfusion volume. Hemoglobin levels rose to an average of at least 11.0 g/dL.
The Marketing Authorization Application (MAA)for exagamglogene autotemcel is scheduled to be sent to the European Medicines Agency (EMA) by the end of 2022.