Highlights
Chronic hepatitis D is an infectious disease caused by hepatitis D virus (HDV), a small spherical enveloped particle that has similarities to viroids and virusoids.
Hepatitis D virus, also called hepatitis delta virus, is a subviral agent (satellite virus) because its assembly, replication, and spread occur only in the presence of hepatitis B virus (HBV).
Infection with HDV can occur simultaneously with HBV infection (coinfection) or in the background of having chronic HBV infection (superinfection).
An estimated 5% of carriers of hepatitis B surface antigen (HBsAg) are coinfected with HDV, which means that the number of HDV carriers is approximately 10–20 million people worldwide. [1]
The simultaneous presence of HBV and HDV leads to more severe symptoms and a more severe course of the disease. Coinfection with HDV and HBV usually manifests spontaneously resolving acute hepatitis, although it can be severe and with an increased risk of developing fulminant hepatitis. Typical prognosis in HDV superinfection with chronic hepatitis B: severe acute hepatitis, chronic hepatitis D in 90% of cases, progression to cirrhosis within 5–10 years in 60%–70% of patients, increased risk of hepatocellular carcinoma (liver cancer). [1] [2] [3]
Treatment of chronic hepatitis D involves treatment of chronic hepatitis B, because the life cycle of HDV is directly dependent on HBV. Pegylated interferon alfa-2a can help in the treatment of chronic hepatitis D, while the monotherapy with antiviral drugs for chronic hepatitis B, such as nucleoside or nucleotide analogues, has little effect on HDV. In cases of fulminant hepatitis or end-stage liver disease due to HDV, orthotopic liver transplantation is the only treatment, after which, however, the risk of HDV/HBV reinfection still remains, and therefore lifelong antiviral therapy is indicated. [1] [2] [4] [5] [6]
The only specialized antiviral drug against HDV (and simultaneously HBV) is bulevirtide. At the end of November 2019, Russia became the first country to approve this drug under the brand name Myrcludex B. At the end of July 2020, bulevirtide received marketing authorization in Europe under the brand name Hepcludex; the authorization is conditional, which means that the drug has yet to finally prove its therapeutic efficacy.
There is a definite difference in the indications for the use of bulevirtide. In Russia, Myrcludex B is indicated for treatment of chronic hepatitis B with delta agent (chronic hepatitis D) in adult patients. In Europe Hepcludex is indicated for treatment of chronic hepatitis D in adults with the presence of its RNA in plasma (or serum) and compensated liver disease.
At the end of November 2021, the New Drug Application (NDA) of bulevirtide was sent to the U.S. Food and Drug Administration (FDA). If the American regulator’s decision is positive, the drug will appear in the U.S. in 2022.
Mechanism of Action of Bulevirtide
Hepatitis D virus (HDV), like hepatitis B virus (HBV), infects hepatocytes by attaching to the cell surface with heparan sulfate proteoglycans (HSPGs) present on it. The Na+-taurocholate cotransporting polypeptide (NTCP, SLC10A1) expressed on the basolateral membrane of differentiated hepatocytes is used for internalization. NTCP, which under normal conditions is responsible for reabsorption of conjugated bile salts into hepatocytes within the enterohepatic circulation, is a highly specific functional receptor for HBV. HDV is then released into the cytoplasm of hepatocytes and its genome is transported to the nucleus; HBV plays no role in the subsequent steps of HDV replication.
HDV requires HBV to recognize NTCP, more specifically the NTCP-binding domain, which is present on the HBV large surface antigen (L-HBsAg) as part of the outer envelope of the HBV supercapsid.
Subcutaneously administered bulevirtide (915207G) is a synthetic stable lipopeptide derived from the PreS1 domain of L-HBsAg. Bulevirtide competes with the latter for binding to NTCP. Blocking NTCP causes HDV and HBV to lose the ability to infect healthy hepatocytes, the viral spreading stops. [1] [2] [3] [4] [5] [6]
Bulevirtide belongs to a new class of entry inhibitors. [7] [8]
Bulevirtide has been shown to reduce viral load along with decreased inflammation and cytolytic processes in the liver, resulting in slower progression of fibrosis and cirrhosis and attenuating the risk of associated complications. Bulevirtide promotes prevention of reinfection of healthy hepatocytes with HDV/HBV along with enhancement of immunological response for clearance of infected hepatocytes. Blocking NTCP over the long term could theoretically pave the way for HDV/HBV elimination.
Bulevirtide was developed by Germany’s MYR Pharmaceuticals, which with the support of Russia’s Hepatera performed the clinical trials necessary for marketing approval in Russia and Europe.
In March 2021, Gilead Sciences, which specializes in treating infectious diseases, including viral hepatitis, acquired MYR for 1.15 billion euros ($1.4 billion) in cash and has pledged up to another 300 million euros ($365 million) if bulevirtide receives approval in the United States.
Bulevirtide: Clinical Efficacy and Safety in Chronic Hepatitis D Treatment
For approval of bulevertide in the U.S. and full marketing authorization in Europe, as much clinical evidence as possible must be gathered to reliably confirm its efficacy and safety.
MYR301
The ongoing MYR301 (NCT03852719) phase 3 (randomized, open-label, multicenter, international) clinical trial involving adult patients (n=150) with chronic hepatitis D with or without cirrhosis is testing the long-term efficacy and safety of bulevirtide with follow-up in an effort to clarify the duration of maintenance of HDV suppression status without any therapy.
Main patient characteristics: mean age 42 years, male 57%, cirrhosis in 47%, previous interferon therapy in 56%, concomitant treatment of chronic hepatitis B with nucleoside/nucleotide analogues in 60%.
Subjects either receive daily subcutaneous injections of bulevirtide at a dose of 2 mg or 10 mg for 144 weeks, or wait 48 weeks and then receive bulevirtide at 10 mg daily for 96 weeks.
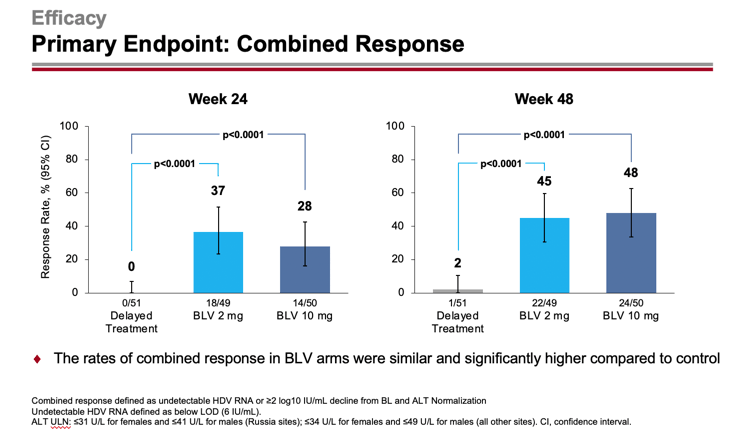
The primary efficacy endpoint of chronic hepatitis D treatment was set by the proportion of patients who demonstrated a combined response: first, an HDV RNA level undetectable by high-precision PCR or its decrease by at least 2 log10 IU/ml (100-fold reduction) from baseline and, second, ALT normalization.
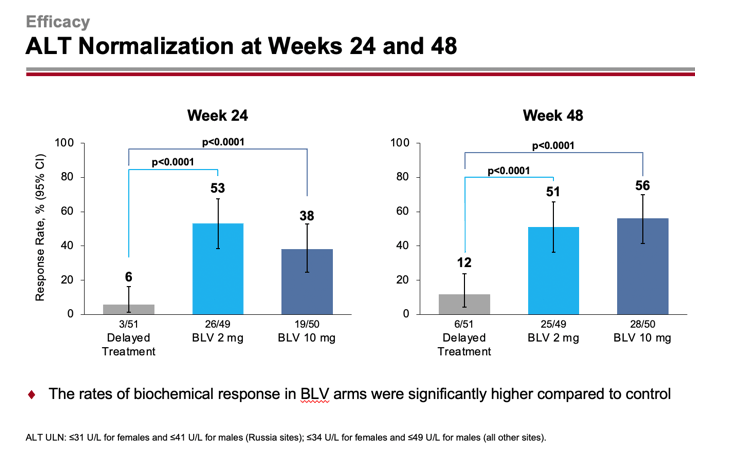
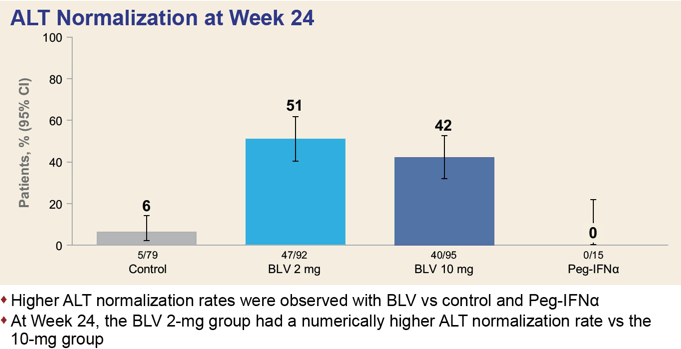
After 24/48 weeks, 37%/45% and 28%/48% of patients in the 2-mg and 10-mg dose groups of Hepcludex reached the primary endpoint — versus 0%/2% of subjects who received no treatment (p<0.0001). Thus, prolonged treatment leads to improved outcomes over time.
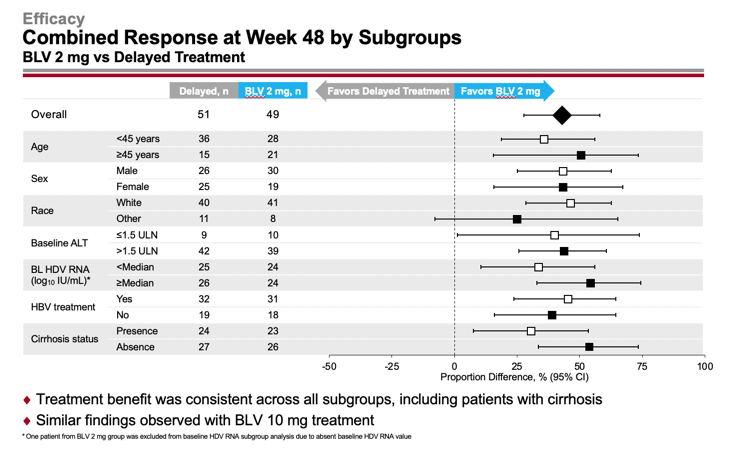
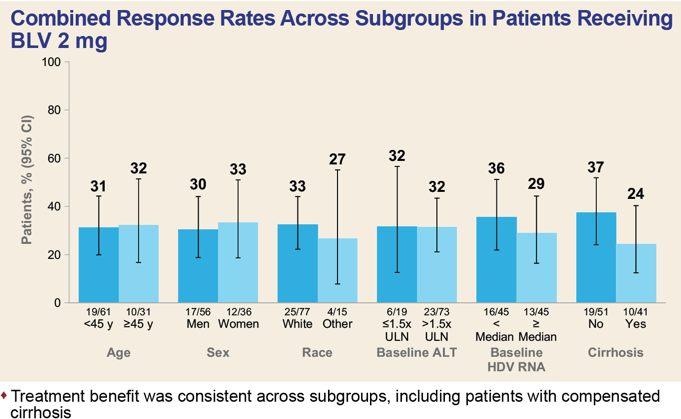
The efficacy of Hepcludex treatment of chronic hepatitis D was generally independent of demographic factors and disease characteristics, such as age, sex, race, baseline ALT and HDV RNA levels, concomitant treatment of chronic hepatitis B, and presence or absence of cirrhosis.
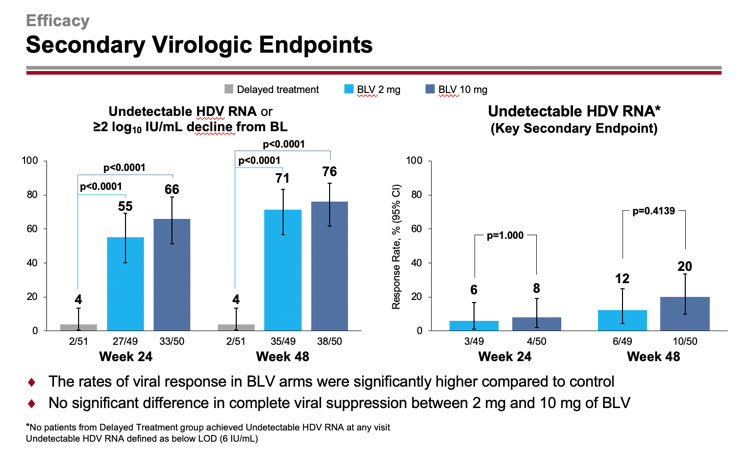
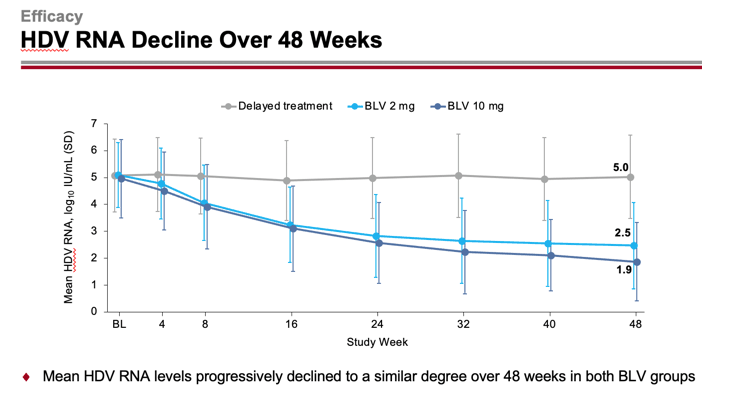
An undetectable level of HDV RNA or a decrease in its level by at least 2 log10 IU/ml from baseline was recorded for 55%/71% and 66%/76% of patients in the 2-mg and 10-mg dose groups of bulevirtide — versus 4%/4% in the group of patients who did not receive any treatment (p<0.0001). At the same time absence of detectable HDV RNA level, i.e. functional cure of chronic hepatitis D, was noted in 6%/12% and 8%/20% of subjects.
Normalization of ALT level was registered for 53%/51% and 38%/56% of patients — versus 6%/12%.
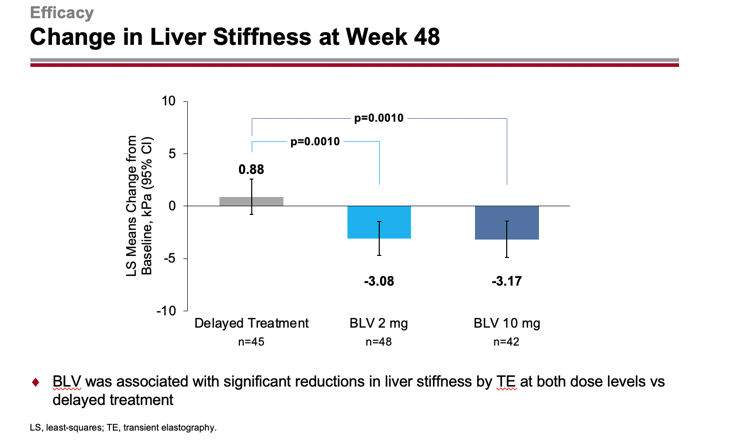
After 48 weeks, liver stiffness according to transient elastography (FibroScan) improved by −3.08 and −3.17 kPa — versus deteriorating by +0.88 kPa (p=0.0010).
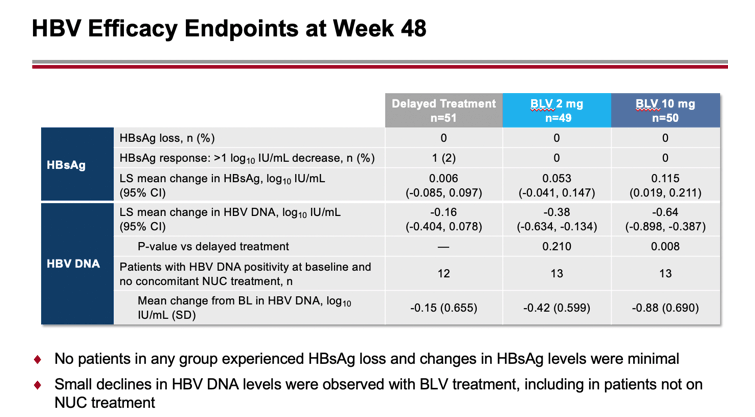
Administration of bulevirtide did not result in loss of HBV surface antigen (HBsAg) or changes in its level were minimal. There was a slight decrease in HBV DNA levels, including among patients who did not receive concomitant treatment for chronic hepatitis B.
Bulevirtide was characterized by acceptable tolerability. There were no serious adverse events related to bulevirtide or those that resulted in discontinuation of treatment. Asymptomatic increases in total bile acids and eosinophils have been reported in the drug groups. Among the most common adverse events to bulevirtide administration were injection site reactions, headache, itching, fatigue, nausea, and dizziness.
MYR204
The ongoing MYR204 (NCT03852433) phase 2b (randomized, open-label, active-controlled, multicenter, international) clinical trial involving adult patients (n=175) focused on the goal of functional cure of chronic hepatitis D by combining bulevirtide with pegylated interferon alfa-2a (this combination exhibits reliable synergistic action) followed by bulevirtide alone to maintain remission status.
Main characteristics of patients: mean age 41 years, male 72%, cirrhosis in 34%.
The combination therapy groups receive daily bulevirtide (2 mg or 10 mg) for 48 weeks along with weekly peginterferon alfa-2a (180 mcg), then bulevirtide alone for 48 weeks, finally followed for another 48 weeks.
The bulevirtide monotherapy group receives it at a dose of 10 mg for 96 weeks with follow-up for 48 weeks.
The control group receives 180 mcg of peginterferon alfa-2a weekly for 48 weeks with follow-up for 48 weeks.
The primary efficacy endpoint for treatment of chronic hepatitis D is determined by the proportion of patients who have demonstrated a sustained virological response (SVR), which is defined as an undetectable HDV RNA level by high-precision PCR at 24 weeks after completion of treatment.
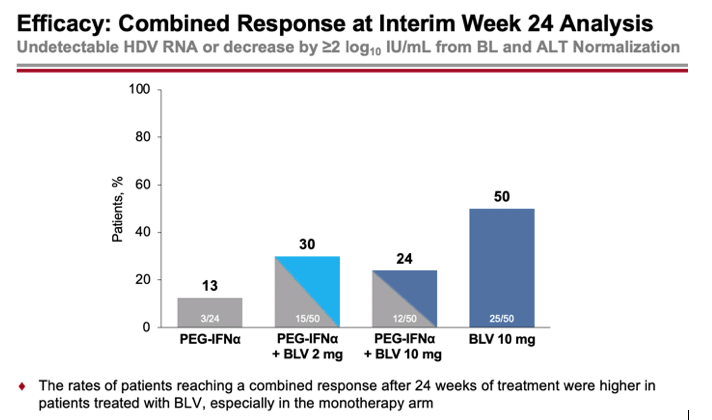
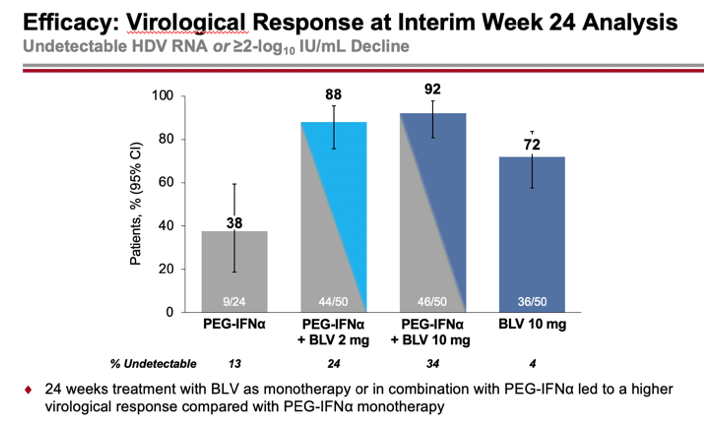
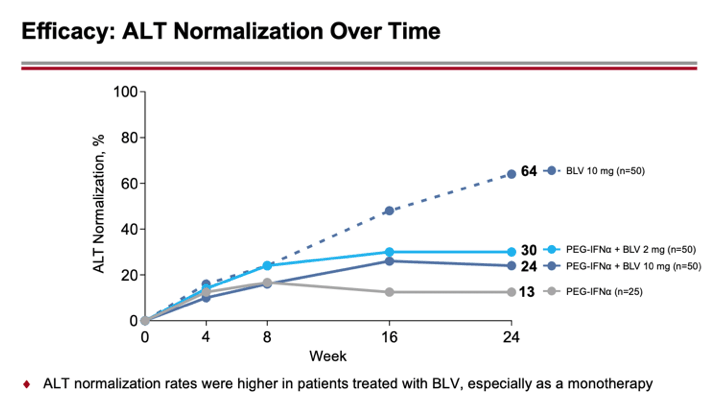
According to interim data collected after 24 weeks of treatment, the percentages of patients who had a combined virological response (undetectable HDV RNA level or decrease by at least 2 log10 IU/ml) and biochemical response (ALT normalization) were 30%, 24%, and 50% in the combined treatment (2 mg or 10 mg Hepcludex on interferon therapy) and 10 mg bulevirtide monotherapy groups — versus 13% in the interferon therapy group.
A virological response was shown by 88%, 92%, and 72% of subjects — versus 38% of participants. The reduction in HDV RNA levels was a median of 3.78, 4.11, and 2.68 log10 IU/ml — versus a 2.01 log10 IU/ml reduction in the control group.
Biochemical response was recorded in 30%, 24%, and 64% — versus 13%.
MYR301, MYR203, MYR202
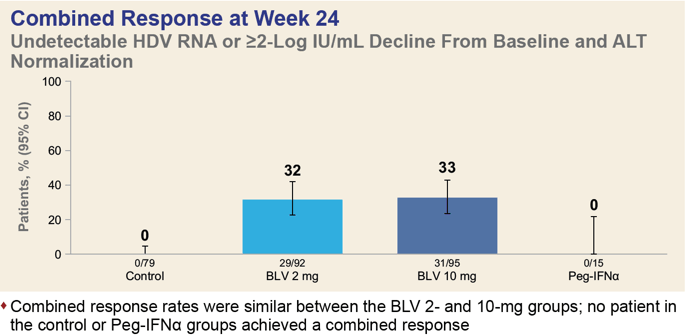
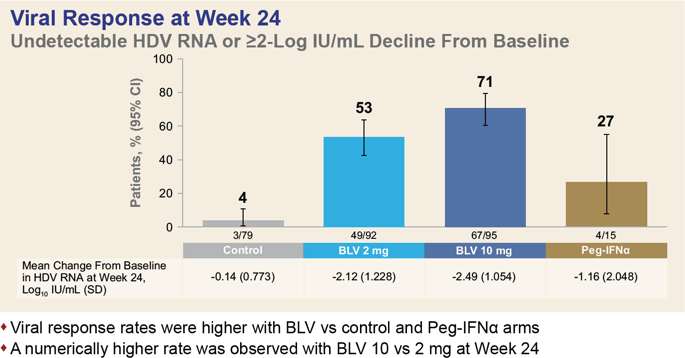
A pooled analysis of data collected in the MYR301 (NCT03852719) phase 3, MYR203 (NCT02888106) phase 2 and MYR202 (NCT03546621) phase 2 clinical trials among adult patients (n=291) with chronic hepatitis D found that after 24 weeks of daily administration of bulevirtide at a dose of 2 mg or 10 mg, one-third of patients, 32% and 33% of subjects, respectively, reached a combined response (HDV RNA level undetectable by high-precision PCR or at least 2 log10 IU/ml reduction and ALT normalization).
A virological response was recorded in 53% and 71% of participants, and a biochemical response in 51% and 42%.
Resistance to Bulevirtide
In clinical trials MYR301 (NCT03852719) and MYR204 (NCT03852433), some patients receiving bulevirtide were classified as virologically non-responders: HDV RNA levels decreased by less than 1 log10 IU/mL over 24 weeks of therapy. In vitro resistance testing and sufficient plasma concentrations of bulevirtide confirmed that the lack of response to treatment was not due to the development of drug resistance to bulevirtide and therefore must be explained by other factors.
Real Clinical Practice #1
Austrian experts studied the effectiveness of treatment of chronic hepatitis D in adult patients (n=23) who received Hepcludex monotherapy (mainly 2 mg/day) for 24 weeks. In case of absence of virological response (reduction of HDV RNA level by at least 2 log10 IU/ml), peginterferon alfa-2a was added to the treatment.
Patients’ baseline characteristics included mean age 48 years, female 57%, 70% had cirrhosis, 91% had received concomitant antiviral therapy with nucleoside/nucleotide analogues, and 78% had not previously responded to interferon therapy.
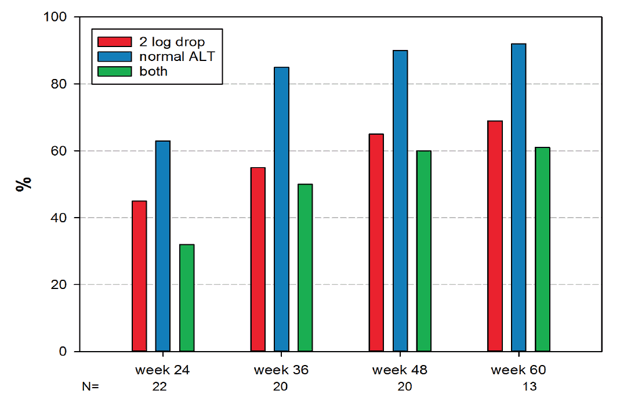
After 24, 36, 48, and 60 weeks of treatment, virological response was seen in 45%, 55%, 65%, and 69% of patients. Biochemical response (normalization of ALT level) was registered in 64%, 85%, 90% and 92% of patients. Combined response (virological and biochemical responses) was recorded in 32%, 50%, 60% and 62% of patients.
The experts made the following conclusions, which help to better understand the place and role of bulevirtide in the clinical practice of treatment of chronic hepatitis D:
- The use of a surrogate endpoint represented by a combined response implies that HDV RNA may still be detectable in a number of patients, while the impact of incomplete HDV suppression on further disease progression remains unknown. In addition, this approach to assessing treatment efficacy is problematic in patients with a low baseline viral load and low or normal ALT levels. The latter is an uncertain marker of liver disease and may be within normal limits even in patients with advanced chronic liver disease.
- The optimal dose of bulevirtide is still unclear, it is possible that doses above 2 mg/day may be more effective.
- The duration of Hepcludex administration is still unknown, more than 2 years of therapy might be necessary to achieve complete viral suppression. For example, two patients whose treatment was discontinued after sustained viral suppression relapsed, although one patient did so after more than 1 year. This is probably due to the persistence of HBsAg, which allows the formation of new HDV particles. On the other hand, spontaneous elimination of HDV may occur without HBV clearance, raising new questions about the mechanism of HDV elimination.
- Additional interferon therapy to bulevirtide seems to add to the chances of successful treatment of chronic hepatitis D, although the unambiguity of this conclusion is debatable. As for adding nucleoside/nucleotide analogues, the therapeutic benefit is likely to be small.
Real Clinical Practice #2
An ongoing study in France of the treatment of chronic hepatitis D with Hepcludex in the observational clinical trial BuleDelta (NCT04166266) revealed the following interim results of 48-week therapy in adult patients (n=55) with either compensated cirrhosis, or severe liver fibrosis (F4 or F3), or moderate fibrosis (F2) and persistently elevated ALT levels.
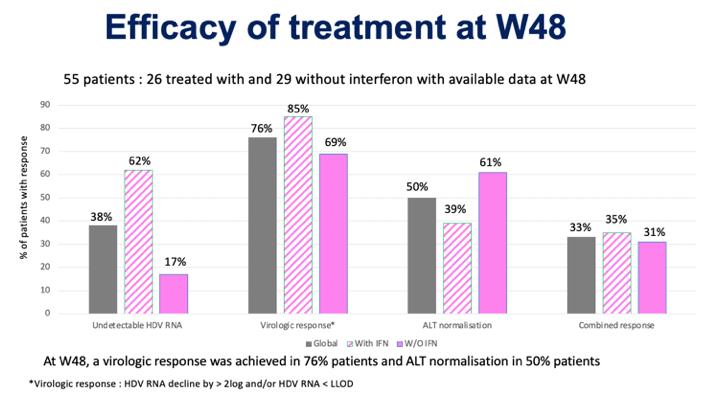
A virological response (HDV RNA level undetectable by high-precision PCR or at least 2 log10 IU/ml reduction) was demonstrated in 85% of patients treated with bulevirtide combined with peginterferon alfa-2a and in 69% of patients treated with bulevirtide alone. At the same time, the absence of HDV RNA was detected in 62% and 17% of patients.
The picture with the biochemical response (normalization of ALT) is different: in 39% and 61% of patients, respectively.
The combined response, combining virological and biochemical responses simultaneously, was fair in 35% and 31% of patients.
Expert Comments
Although bulevirtide is approved in Russia and Europe, it is currently not known exactly how long and how exactly one will have to be treated. Bulevirtide may have to be taken for several years or even for life to achieve a sustained remission of chronic hepatitis D in order to avoid a relapse. Treatment will most likely have to be organized on a 48-week (or longer) course of pegylated interferon alfa-2a, and then adhere to maintenance therapy with bulivertide alone (or its combination with nucleoside/nucleotide analogues for chronic hepatitis B). However, combination therapy with bulivertide and peginterferon or nucleoside/nucleotide analogues is still poorly understood. Again, bulivertide is not a cure-all because it does not help everyone with chronic hepatitis D.
Overall, the regulatory approval of bulevitide was made hastily: An experimental drug has actually entered the market, leaving open a number of important questions that are gradually being addressed as the clinical data accumulate. This is quite acceptable given the categorical absence of specialized drugs against chronic hepatitis D.
The MYR301 (NCT03852719) phase 3 clinical trial should confirm the validity of long-term prescribing of bulivertide in terms of viral suppression.
Much more important for patients is the MYR204 (NCT03852433) phase 2b clinical trial designed to prove that bulevitide in combination with interferon therapy can functionally cure chronic hepatitis D.
The prerequisites for a successful cure do exist, just look at the results of the preceding clinical trial MYR203 (NCT02888106) phase 2b (randomized, open-label, active-controlled, multicenter), whose data formed the basis of the NDAs for bulevirtide for Russia and Europe. You should know that the trial was conducted exclusively in Russia, which makes the community of doctors and patients wary because of the impossibility of absolutely excluding risks of negligence in any clinical trials. In addition, the small sample size of patients (the trial was completed by 73 patients) cannot serve as unequivocally sufficient proof of treatment efficacy.
Adult patients (n=90) with chronic viral hepatitis D were divided into six groups, who received the following treatment for 48 weeks: peginterferon alfa-2a [group A, control], bulevirtide 2 mg plus peginterferon alfa-2a [group B], bulevirtide 5 mg plus peginterferon alfa-2a [group C], bulevirtide 2 mg [group D], bulevirtide 10 mg plus peginterferon alfa-2a [group E], bulevirtide 10 mg (5 mg twice daily) plus tenofovir 300 mg [group F].
Subcutaneous interferon was administered weekly at 180 mcg, subcutaneous bulevirtide and oral tenofovir daily.
The primary efficacy endpoint for treatment of chronic hepatitis D, the stated proportion of patients whose HDV RNA levels decreased to an undetectable high-precision PCR value at 24 weeks after completion of therapy, was 0% (95% CI: 0.0 to 21.8), 53% (95% CI: 26.6 to 78.7), 27% (95% CI: 7.8 to 55.1), 7% (95% CI: 0.2 to 31.9), 7% (95% CI: 0.2 to 31.9), and 33% (95% CI: 11.8 to 61.6) of subjects.
Only groups A and F showed a statistically significant difference with the control group (p=0.0022 and p=0.0421), with the combination of 2-mg bulevirtide and interferon being the most effective, providing functional cure of chronic hepatitis D in half of the patients.
- A certain oddity can be noted in the peginterferon alfa-2a monotherapy group, which had no effect at all on HDV RNA clearance, while in other clinical trials interferon therapy for chronic hepatitis D proved its worth. Thus, a sustained virological response (absence of HDV RNA 24 weeks after at least 48 weeks of interferon therapy) was observed in 17% [1], 43% [2], 18% [3], 28% [4], 29% [5] of patients. It should be understood that interferon therapy, even if it provides a sustained virological response, is not a reliable guarantee that there is no risk of recurrence of chronic hepatitis D, which over half of patients may face in the future. [6] It is possible that peginterferon alfa-2a should be prescribed for much longer. [7] [8] [9] [10]
The same was true for normalization of ALT levels (≤ 31 U/L for women and ≤ 41 U/L for men): in 10% (95% CI: 0.3 to 44.5), 54% (95% CI: 25.1 to 80.8), 33% (95% CI: 11.8 to 61.6), 23% (95% CI: 5.0 to 53.8), 36% (95% CI: 12.08 to 64.9), and 36% (95% CI: 12.8 to 64.9) of patients.
A combined treatment response combining absence of HDV RNA and normalization of ALT was reported for 0% (95% CI: 0.0 to 21.8), 47% (95% CI: 21.3 to 73.4), 13% (95% CI: 1.7 to 40.5), 7% (95% CI: 0.2 to 31.9), 7% (95% CI: 0.2 to 31.9) and 13% (95% CI: 1.7 to 40.5) of participants.
In terms of functional cure of chronic hepatitis B, which refers to clearance of HBV surface antigen (HBsAg) to undetectable levels with or without associated antibody seroconversion (anti-HBs), this status was achieved in 23% (95% CI: 5.0 to 53.8) and 8% (95% CI: 0.2 to 36.0) of patients treated with a combination of 2-mg bulevirtide on interferon therapy, who were in status with and without anti-HBs, respectively.