Highlights
Cresomycin is a new oral antibiotic that can overcome diverse forms of antimicrobial resistance (AMR), rendering current antibacterial drugs ineffective [1].
In vitro and in vivo, cresomycin is effective against Gram-positive and Gram-negative microorganisms, including multidrug-resistant (MDR) strains of bacteria such as Staphylococcus aureus, Escherichia coli, Pseudomonas aeruginosa, Acinetobacter baumannii, Neisseria gonorrhoeae, and others. Cresomycin is effective in treating infections of the skin, soft tissues, intestines, urinary and genitourinary tracts, lungs, blood, and other organs and tissues.
Cresomycin exhibits bacteriostatic activity against S. aureus.
In vitro experiments on human cells indicated low cytotoxicity of cresomycin. Therapeutic doses of cresomycin did not cause hemolysis of erythrocytes.
Cresomycin inhibits protein synthesis in microorganisms by binding to the active site of the bacterial ribosome.
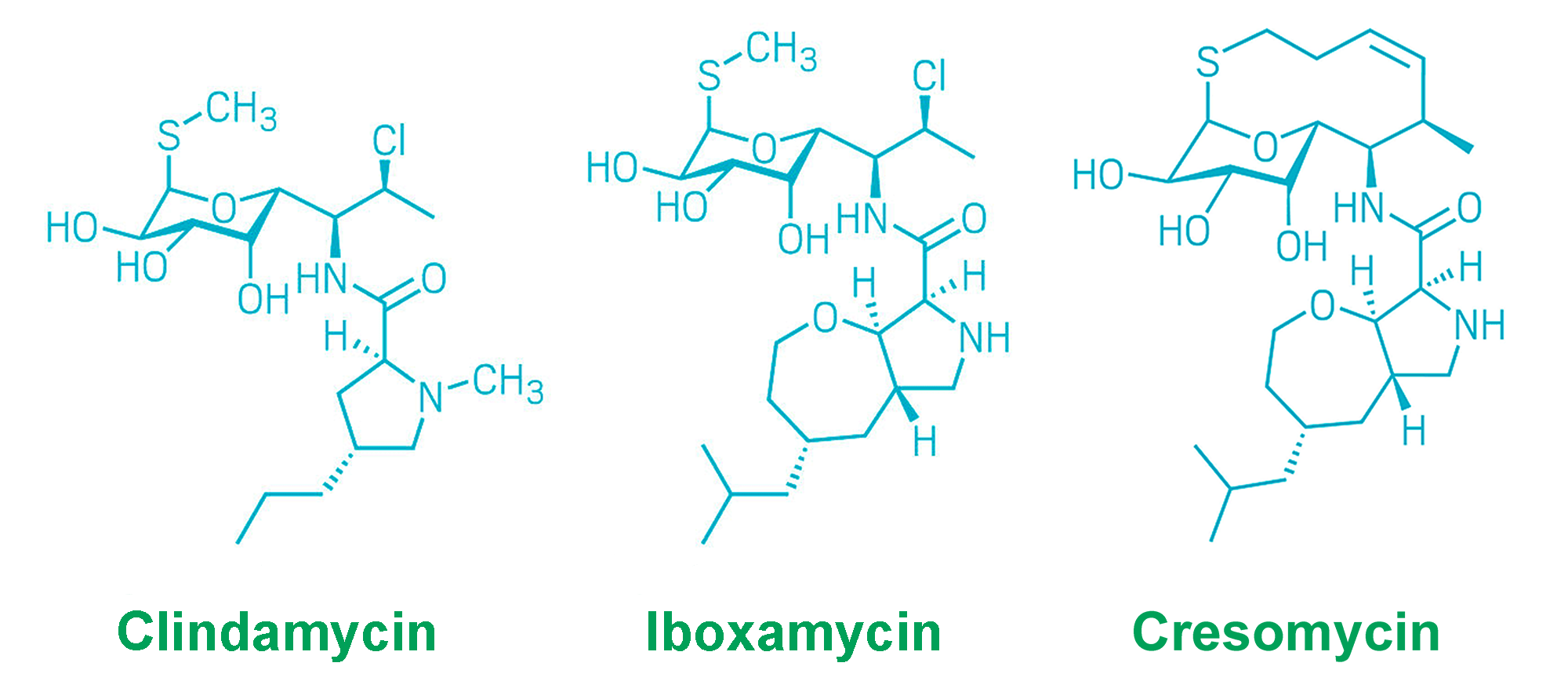
Cresomycin belongs to the bridged macrobicyclic oxepanoprolinamide antibiotics, which are similar to lincosamides.
Cresomycin was developed using a technique called component synthesis, which involves pre-creating the individual parts of drug compounds and then assembling them into a single molecule.
The discovery of cresomycin is significant due to the increasing number of deaths caused by AMR infections. In 2019, AMR infections caused 1.27 million deaths, surpassing the number of deaths caused by HIV/AIDS (864,000) and malaria (643,000) [2]. It is projected that AMR infections will cause 10 million deaths annually by 2050 [3].
Activity
Cresomycin is effective against antibiotic resistance mechanisms provided by the bacterial genes Erm and Cfr in Gram-positive bacteria. Erm confers resistance to macrolide, lincosamide, and streptogramine group B antibiotics, while Cfr confers resistance to phenicols, lincosamides, oxazolidinones, pleuromutilins, and streptogramine group A antibiotics. Cresomycin also bypasses antibiotic resistance realized by other bacterial genes, including lnuA, lsaA, and cplR.
Cresomycin overcomes antibiotic resistance in Gram-negative bacteria due to their outer membrane.
In vitro, cresomycin has demonstrated proper antibacterial activity against the microorganisms listed in reference [1], as determined by the minimum inhibitory concentration (MIC):
Gram-positive bacteria
- Staphylococcus: S. aureus, S. haemolyticus, S. epidermidis
- Streptococcus: S. pneumoniae, S. pyogenes
- Enterococcus: E. faecalis, E. faecium, E. casseliflavus, E. gallinarum
- Clostridium: C. difficile.
Gram-negative bacteria
- Neisseria: N. gonorrhoeae
- Escherichia: E. coli
- Klebsiella: K. pneumoniae
- Acinetobacter: A. baumannii
- Pseudomonas: P. aeruginosa
- Moraxella: M. catarrhalis
- Haemophilus: H. influenzae.
The fact that certain bacteria are not listed does not necessarily imply that cresomycin is ineffective against them. It is important to note that this antibiotic has not yet undergone extensive microbiological testing on a wide range of microorganisms.
In mouse models of sepsis caused by S. aureus, cresomycin provided a 100% survival rate. In mouse models of neutropenic femoral infection, administration of cresomycin reduced the bacterial burden of Cfr-expressing S. aureus, ErmA-expressing S. aureus, and carbapenem-resistant E. coli and P. aeruginosa by −4.6, −2.2, −2.6, and −2.7 log10 CFU, respectively.
Development
The development of cresomycin was led by Andrew Myers from Harvard University and Yury Polikanov from the University of Illinois at Chicago.
In 2021, scientists reported on iboxamycin, a new antibiotic. It belongs to the group of lincosamides and retains half of the chemical structure of clindamycin. The other half is modified to bind more closely to the target in the active site of the bacterial ribosome. This modification results in the microorganism reliably losing the ability to synthesize proteins [1].
In the computational study of iboxamycin’s conformational structure, the modification of the site borrowed from clindamycin was tested to create a macrocycle that would be fixed in the most favorable position for bacterial ribosome binding. The experiments revealed that a ten-membered ring is the optimal choice.
Further studies in structural biology have revealed that cresomycin binds strongly to several bacterial ribosomes, including drug-resistant ones. It is surprising to see how the molecule overcomes resistance: in ribosomes, cresomycin actually repels the methyl groups that prevent binding. This is most likely because cresomycin binds very tightly to the ribosome since it is “pre-organized”, meaning it does not have to expend as much energy as existing antibiotics to align itself with the target [2] [3].
This study challenges the conventional approach to developing new antibiotics. It suggests that structural changes to existing antibacterial drugs do not necessarily have to target the region in the ribosome responsible for resistance. The research indicates that when the drug has a high enough affinity for its target, such as the catalytic peptidyl transferase center (PTC) of the ribosome, it can displace the methylated nucleotide residue responsible for antibiotic resistance [4].
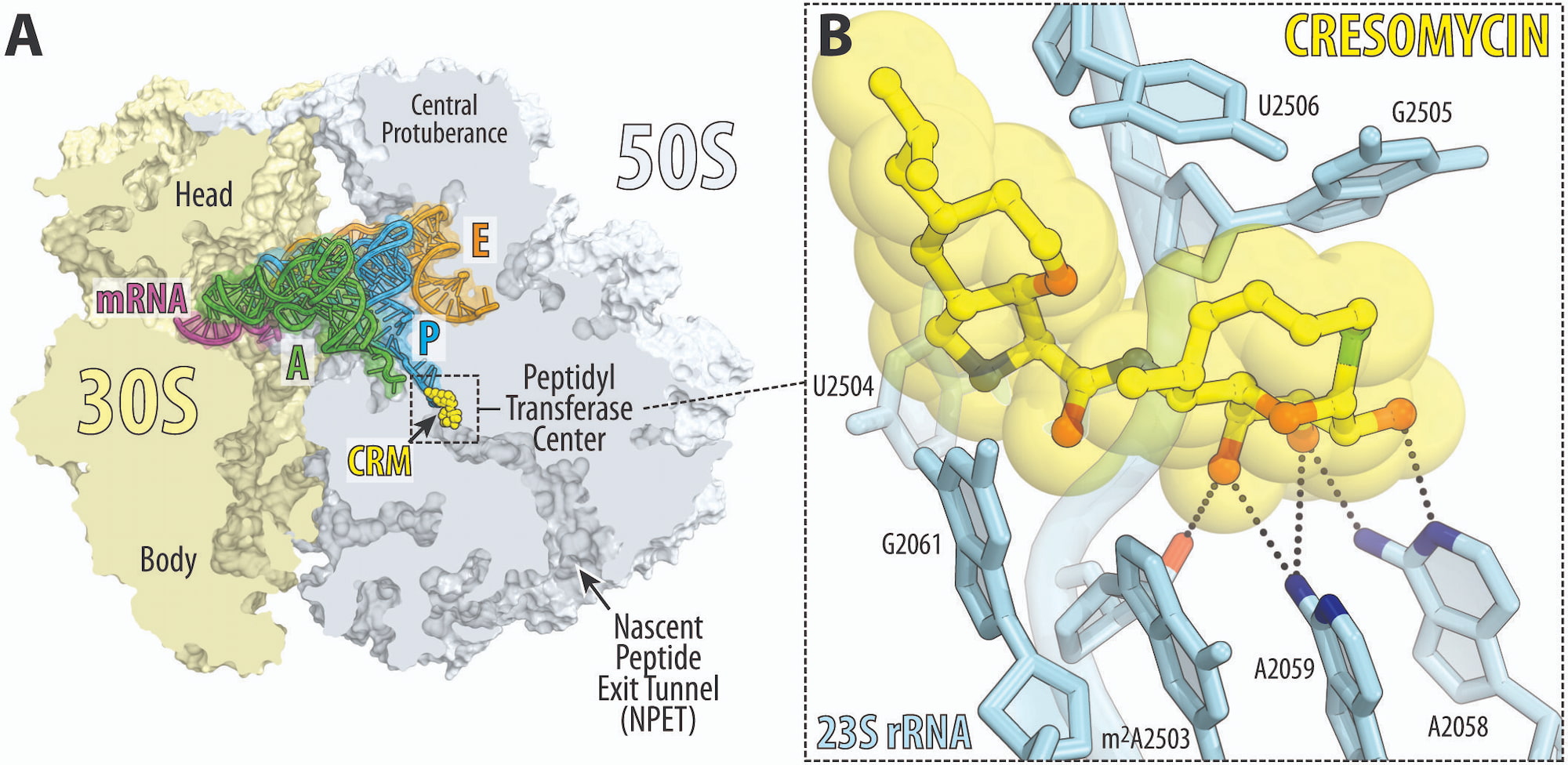
An appropriate analogy is the following: Antibiotic resistance that targets the bacterial ribosome develops through the expression of genes that produce ribosomal RNA methyltransferases. These enzymes insert a methyl group into the ribosome, which repels the antibiotic when it tries to bind to the ribosome. The methyl group acts as a tiny pushpin attached to the site where the antibiotic hopes to bind. But cresomycin binds so tightly and strongly to the ribosome that it essentially drives that pushpin inward [5].
In reality, it’s much more complicated than that. According to the research of Myers and Polikanov, X-ray crystallography, which allowed ribosomes to be visualized with near-atomic precision, revealed two of their defense tactics. The methyl group not only creates a direct steric hindrance that physically blocks binding, but also induces a conformational change in the binding site. Iboxamycin and cresomycin, apparently, displace the methyl group from its canonical position, depriving the ribosome of its ability to resist drugs [6].


Without looking back at the promising future of cresomycin, more data need to be collected on its efficacy against Gram-negative bacteria, which are protected by the outer membrane and are the main problem of the global burden of antibiotic resistance. However, the synthetic design of cresomycin is elegant and represents an exciting opportunity for further development [7].
The study is of great significance. First, it has increased academic interest in antibiotic development. Second, cresomycin not only inhibits the synthesis of bacterial proteins, but also suppresses the bacteria themselves, including resistant strains.
“There are a lot of compounds that can inhibit protein synthesis. Very few of those will inhibit bacteria. Out of those few, very few will be able to kill bacteria inside a live animal.“And almost none will be able to kill multidrug-resistant bacteria in an animal.”
Yury Polikanov [8].
In February 2024, the Myers lab was awarded a $1.2 million grant by CARB-X, a global non-profit partnership that accelerates antibacterial products to address drug-resistant bacteria. The grant will be used for preclinical studies of cresomycin and other antibacterial compounds [9].
It is possible that, in the future, researchers may synthesize an antibacterial candidate more suitable than cresomycin for further clinical development. However, it is already evident that the novelty of cresomycin is impressive. According to expert estimates, only about one in a thousand experimental antibiotics reaches the level of success that cresomycin has achieved. Of the three or four dozen that have reached the level of cresomycin, only one will be fortunate enough to receive regulatory approval [10].
Background
In the spring of 2016, the American public was alarmed to learn of the first known case of infection with a bacteria resistant to colistin, a last-resort antibiotic. Superbug Escherichia coli carrying the mcr-1 gene was found in a 49-year-old woman suffering from a urinary tract infection [1].
Thomas Frieden, director of the U.S. Centers for Disease Control and Prevention (CDC), lamented that “it is the end of the road for antibiotics unless we act urgently.” [2]
Pharmaceutical companies often avoid developing new antibiotics due to the lack of profitability in recouping the costs of creating them. Instead, they tend to focus on drugs for chronic diseases, which are more profitable [3].
Meanwhile, Andrew Myers has developed a technology platform to expedite the development of macrolide antibiotics. These popular antimicrobials are based on a large macrocyclic lactone ring and are among the safest antibiotics. However, macrolides becoming increasingly ineffective due to bacterial resistance [4].
Myers’ method, which was described in May 2016, involves using simple and commercially available chemicals (or their derivatives) to create building blocks. These simple building blocks are then further assembled into new ring-shaped molecules, which are tested for their ability to fight pathogens. More than 350 compounds were synthesized using this method, most of which had antimicrobial activity. Two of these compounds were able to cope with clinical isolates characterized by the most complex combinations of antibiotic resistance mechanisms [5].
In the post-penicillin era, pharmaceutical companies scoured the world for natural compounds that could serve as antibiotics. In 1949, erythromycin, the first macrolide, was discovered in a soil sample in the Philippines. At an unprecedented of rate, it was approved for human use in 1952. Subsequently, antibiotic researchers often began with the erythromycin molecule, using a process called semisynthesis, where the original chemical structure was modified, but not significantly. Semisynthesis dominated for many decades [6].
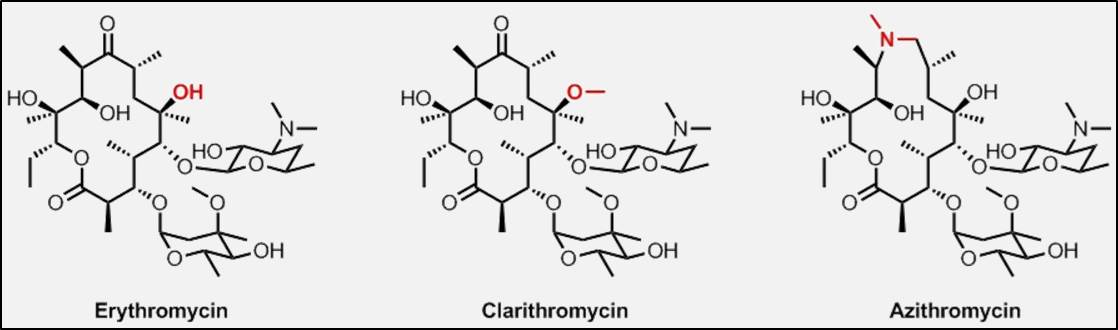
Myers’ practical approach to antibiotic development is fully synthetic, addressing the convergent assembly of molecular structures from simple chemical building blocks or modules. This approach eliminates the need for chemical modification of structurally complex fermentation products, reducing the number of stages required to create a new molecule, increasing the yield of the finished product, and allowing the assembly of molecules with structures that are inaccessible to semisynthesis.
The construction of new molecules through modularity eliminates the difficulties associated with working with erythromycin, a complex natural compound. Synthesizing from scratch allows for the modification of any part of the molecule without affecting others. This is because it is not easy to carry out the chemical process so that it affects only the necessary part of the structure without changing others.
Examples of synthesizing structural analogs of telithromycin and solithromycin, as well as a hybrid scaffold combining structural features of azithromycin and solithromycin, were demonstrated.
It should be noted that funding for the research was difficult. Due to the lack of income from the U.S. National Institutes of Health (NIH), it was necessary to ask for help from private investors. In 2013, the Gustavus and Louise Pfeiffer Research Foundation, the Blavatnik Biomedical Accelerator at Harvard University, and some Harvard alumni found not too much money [7].

In March 2015, Myers et al. founded Macrolide Pharmaceuticals to commercialize their scientific research. The Harvard Office of Technology Development licensed their patent-protected intellectual property to the company [8].
Macrolide immediately received a deposit of $22 million from venture capitalists. In early March 2018, CARB-X pledged up to $6.81 million in funding. In early April of that year, investors added $20 million to the company’s piggy bank [9] [10] [11].
However, in 2019, the business was reformatted and Macrolide had ceased to exist. Zikani Therapeutics was established to develop oral macrolide-based Ribosome Modulating Agents (RMAs) synthesized on the proprietary TURBO-ZM platform for the treatment of rare diseases caused by nonsense mutations, such as cystic fibrosis with CFTR class I mutations, familial adenomatous polyposis (FAP), colorectal cancer with APC mutation, and recessive dystrophic epidermolysis bullosa (RDEB). In April 2021, Eloxx Pharmaceuticals acquired Zikani [12].
Lessons
Myers’ research replicates his team’s technological approach to developing new and fully synthetic tetracycline antibiotics, which eventually led to the creation of Tetraphase Pharmaceuticals in 2006 [1] [2].
Tetraphase, which became a public company in 2013, developed the injectable antibacterial drug Xerava (eravacycline) for the treatment of complicated intra-abdominal infections (cIAI) and made it available at the end of August 2018. However, the arrival of eravacycline, a fully synthetic fluorocycline, was not sufficient to keep the business afloat [3].
Tetraphase faced very low demand for Xerava, earning only $3.6 million in 2019. First, doctors, doctors reserved eravacycline for severe cases due to concerns about bacterial resistance development. Second, the U.S. Food and Drug Administration (FDA) clearly stated in the prescribing information for eravacycline that it should be used only against bacteria that are sensitive to the drug.
In late July 2020, a couple of years after Xerava’s debut, Tetraphase was forced to sell itself to La Jolla Pharmaceutical for $43 million. Additionally, Tetraphase was promised up to $16 million more if eravacycline sales in the U.S. reached a certain volume. The amount offered did not compare to the market value of the company, which at its peak reached $1.8 billion [4].
Tetraphase’s business decline was preceded by two unsuccessful attempts to treat complicated urinary tract infections (cUTI). Neither the oral nor the original intravenous version of eravacycline performed well. These failures occurred in early September 2015 and mid-February 2018, causing Tetraphase’s stock quotes to drop by 80% and 50% [5] [6].
However, eravacycline did not stay in the new hands for long. At the end of August 2022, again after two years, La Jolla was acquired by the multidisciplinary holding company Innoviva [7].