Клиническая проверка показала, что сочетание препаратов «Опдиво» (Opdivo, ниволумаб) и «Ервой» (Yervoy, ипилимумаб), назначаемое в ходе первоочередного лечения неоперабельного рака печени (гепатоцеллюлярной карциномы), превосходит эффективность, обеспечиваемую терапией в лице препарата «Ленвима» (Lenvima, ленватиниб) или «Нексавар» (Nexavar, сорафениб).
ОСНОВНЫЕ ФАКТЫ
«Бристол-Майерс Сквибб» (Bristol-Myers Squibb) продемонстрировала преимущество сочетания из ниволумаба (nivolumab) и ипилимумаба (ipilimumab), блокаторов PD-1 и CTLA-4, над ленватинибом (lenvatinib) или сорафенибом (sorafenib), тирозинкиназными ингибиторами, продвигаемыми соответственно «Эйсай» (Eisai) / «Мерк и Ко» (Merck & Co.) и «Байер» (Bayer), в ходе перволинейной терапии рака печени.
По отношению к препаратам сравнения иммунноонкологический коктейль снизил риск смерти на 21% и снизил риск прогрессирования заболевания или смерти на 13%.
Этого недостаточно, чтобы опередить нынешний стандарт в лице комбинации из «Тецентрика» (Tecentriq, атезолизумаб) и «Авастина» (Avastin, бевацизумаб) — блокатора PD-L1 и ингибитора VEGF авторства «Рош» (Roche).
Однако в абсолютном исчислении продление общей выживаемости оказалось превосходным.
Регистрационное досье отправлено в адрес регуляторов.
ПРЯМАЯ РЕЧЬ
«Медиана общей выживаемости получилась одной из самых длинных, которые мы когда-либо наблюдали в ходе лечения распространенной гепатоцеллюлярной карциномы. Мы уверены, что разработали новый стандарт лечения».
Питер Галле (Peter Galle), клинический гепатолог из Медицинского центра при Майнцском университете (земля Рейнланд-Пфальц, Германия).
«Отмеченная нами частота уменьшения опухоли — одна из самых высоких среди других вариантов лечения рака печени. Высокий уровень ответа на терапию повышает шансы трансформации заболевания из неоперабельного в поддающееся резекции».
Лаура Гофф (Laura Goff), исполнительный медицинский директор Центра ухода за онкологическими пациентами при Онкологическом центре Вандербильта — Инграма (VICC, шт. Теннесси, США).
«Несмотря на продолжающееся развитие фармакологической науки, прогноз для пациентов с гепатоцеллюлярной карциномой по-прежнему остается плохим. Вот почему важно предложить им новые способы лечения, которые, возможно, помогут».
Дана Уолкер (Dana Walker), вице-президент и руководитель глобальной программы по раку желудочно-кишечного тракта и мочеполовой системы «Бристол-Майерс Сквибб» (Bristol-Myers Squibb).
КЛИНИЧЕСКИЕ ПОДРОБНОСТИ
Клиническое исследование CheckMate 9DW (NCT04039607) фазы III (рандомизированное, открытое, с активным контролем, многоцентровое, международное) пригласило взрослых пациентов (n=668) с распространенной гепатоцеллюлярной карциномой, ранее не проходившей системной терапии.
Участникам назначали либо комбинацию из ниволумаба и ипилимумаба, либо ленватиниб или сорафениб (на выбор исследователя) — до момента прогрессирования заболевания или неприемлемой токсичности.
После наблюдений на протяжении медианных 35,2 месяца (26,8–48,9) результаты получились следующими [1] [2] [3].
Общая выживаемость в группе «Опдиво» с «Ервоем» вышла к 23,7 месяца (95% ДИ [здесь и далее]: 18,8–29,4) — против 20,6 месяца (17,5–22,5) в группе «Ленвимы» или «Нексавара». Риск смерти снизился на относительный 21%: отношение риска (hazard ratio, HR) 0,79 (0,65–0,96); p=0,018.
Вероятность остаться в живых на протяжении 24 месяцев составила 49% против 39%, 36 месяцев — 38% против 24%.
Частота общего ответа (ORR) составила 36% (31–42), включая 7% полных ответов (CR), — против 13% (10–17), в том числе 2% CR (p<0,0001).
Медиана длительности ответа (DoR) получилась равной 30,4 месяца (21,2–NE) — против 12,9 месяца (10,2–31,2).
Медиана выживаемости без прогрессирования (PFS) обозначилась на уровне 9,1 месяца (6,6–10,5) — против 9,2 месяца (7,9–11,1). Риск прогрессирования заболевания или смерти снизился на относительных 13%: HR 0,87 (0,72–1,06). Статус PFS в течение 18 месяцев оказался справедливым для 34% пациентов против 18%, 24 месяцев — 28% против 12%.
Назначение иммуноонкологического коктейля привело к снижению риска ухудшения симптомов заболевания на относительных 24%: HR 0,76 (0,62–0,93); p=0,0059.
КОНТРАРГУМЕНТЫ
Использование ленватиниба или сорафениба в качестве контрольной группы — сомнительный выбор «Бристол-Майерс Сквибб». Разумнее было остановиться на более эффективных схемах, представленных либо сочетанием атезолизумаба (atezolizumab) с бевацизумабом (bevacizumab), за которым стоит «Рош», либо дуэтом «Имфинзи» (Imfinzi, дурвалумаб) с «Имджудо» (Imjudo, тремелимумаб) — блокатора PD-L1 с блокатором CTLA-4, продвигаемым «АстраЗенека» (AstraZeneca). Первая схема является предпочтительной, вторая выступает альтернативной в случае противопоказаний к назначению бевацизумаба или его непереносимости.
Согласно нынешним рекомендациям Американского общества клинической онкологии (ASCO), атезолизумаб с бевацизумабом лидирует по эффективности первоочередного лечения гепатоцеллюлярной карциномы, улучшая выживаемость и сдерживая прогрессирование заболевания относительно сорафениба: OS HR 0,66 (0,52–0,85) и PFS HR 0,65 (0,53–0,81) [1] [2].
Применение дурвалумаба (durvalumab) с тремелимумабом (tremelimumab) характеризуется только улучшением выживаемости относительно сорафениба: OS HR 0,78 (0,67–0,92) и PFS HR 0,90 (0,77–1,05) [3] [4].
ЭКСПЕРТНЫЕ КОММЕНТАРИИ
Начиная с марта 2020 года, «Опдиво» с «Ервоем» применяются во второлинейной терапии гепатоцеллюлярной карциномы. Но бизнес «Бристол-Майерс Сквибб» требует расширения охвата пригодных пациентов.
Результаты клинической проверки этого сочетания в перволинейной терапии рака печени получились на первый взгляд идентичными таковым в случае перволинейного использования дурвалумаба с тремелимумабом. Но есть важные отличия.
Во-первых, «Опдиво» с «Ервоеем» обеспечили более продолжительную общую выживаемость с медианой 20,6 месяца — против 16,4 месяца в случае «Имфинзи» с «Имджудо».
Во-вторых, они повысили шансы остаться в живых: до 49% и 38% на протяжении 24 и 36 месяцев — против 41% и 31%.
В-третьих, на лечение ответила большая пропорция пациентов: ORR 36% и CR 7% — против ORR 20% и CR 3%.
В-четвертых, ответ на лечение продолжался дольше: 30,4 месяца — против 22,3 месяца.
Однако справиться с прогрессированием заболевания удалось аналогично плохо.
Осталось разобраться с нежелательными явлениями и их частотой, чтобы понять, насколько новая схема лечения окажется переносимой.
Перспективность ниволумаба с ипилимумабом в контексте первоочередного лечения гепатоцеллюлярной карциномы сомнений не вызывает, ведь долгосрочные 4-летние наблюдения за пациентами, получавшими дурвалумаб с тремелимумабом, установили прилично высокую пропорцию долгожителей, то есть перешагнувших отметку 36-месячной выживаемости: каждый четвертый (25%) — против 15% в группе сорафениба [1].
В идеале было бы правильным напрямую сравнить эти две схемы с «Тецентриком» и «Авастином», но гранды фармотрасли на такое вряд ли пойдут.
ОДНАКО
Списывать сорафениб или ленватиниб со счетов пока рано. Согласно систематическому обзору и метаанализу, сочетание тирозинкиназного ингибитора, блокатора PD-1 и локорегионарной терапии характеризовалось максимальными шансами на конверсию изначально неоперабельной гепатоцеллюлярной карциномы в поддающуюся радикальной резекции, то есть с потенциалом очень длительной выживаемости [1].
«Санленка» (Sunlenca, ленакапавир) — новый лекарственный препарат, предназначенный для антиретровирусной терапии (АРТ) взрослых пациентов с инфекцией вируса иммунодефицита человека 1 (ВИЧ-1) с множественной лекарственной устойчивостью (МЛУ).
ОСНОВНЫЕ ФАКТЫ
Приблизительно 2% популяции ВИЧ-инфицированных характеризуются мутационной резистентностью вируса к назначению существующих АРТ-препаратов, что делает весьма проблематичной задачу достижения и поддерживания статуса вирусной супрессии.
Ленакапавир (lenacapavir; LEN), разработанный «Гилеад сайенсиз» (Gilead Sciences), — первый представитель нового класса АРТ-препаратов, называемых ингибиторами капсида ВИЧ.
«Санленка» применяется совместно с другими АРТ-препаратами в составе комплексных схем лечения, подбираемых в индивидуальном порядке.
В поддерживающем режиме АРТ-терапии «Санленка» назначается подкожными инъекциями 1 раз в 6 месяцев.
«Санленка» получил маркетинговое разрешение Европейского агентства по лекарственным средствам (EMA) в середине августа 2022 года: ленакапавир показан в том случае, если невозможным иным способом подобрать эффективную супрессивную АРТ-схему [1].
«Санленка» одобрен Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) в конце декабря 2022 года: ленакапавир показан в том случае, если текущая АРТ-схема перестала удовлетворять по причине резистентности, непереносимости или небезопасности [2].
В России «Санленка» не зарегистрирован, соответствующих клинических испытаний не проводится.
ПРЯМАЯ РЕЧЬ
«Ленакапавир, предлагая долгожданный вариант терапии дважды в год, удовлетворит критически важную потребность людей со сложной историей лечения, подверженных большому риску прогрессирования заболевания».
Жан-Мишель Молина (Jean-Michel Molina), заведующий отделением инфекционных болезней в больницах Сен-Луи и Ларибуазьер (Париж, Франция).
«Доступность новых классов антиретровирусных препаратов крайне важна для людей с множественной лекарственной устойчивостью к ВИЧ, не отвечающих на доступную терапию».
«Появление ленакапавира, возможно, поможет сложным пациентам прожить более долгую и здоровую жизнь».
Дебра Бирнкрант (Debra Birnkrant), директор департамента противовирусных препаратов в Центре по оценке и изучению лекарственных препаратов при FDA.
«Ученые „Гилеад“, разработавшие уникальный и мощный антиретровирусный препарат, продолжают трудиться над тем, чтобы кардинально изменить ситуацию на пути к прекращению эпидемии ВИЧ».
Дэниел О’Дэй (Daniel O’Day), председатель правления и исполнительный директор «Гилеад сайенсиз» (Gilead Sciences).
СУТЬ ВОПРОСА
Для подавляющего большинства людей, живущих с инфекцией ВИЧ-1, можно подобрать эффективную АРТ-схему, которая продолжительно и надежно справляется с задачей супрессии (подавления) вируса [1].
Однако в некоторых случаях АРТ-схема сталкивается с неудачей: либо из-за вирусной резистентности (устойчивости) к лекарственным препаратам разных классов, либо по причине неприемлемых нежелательных явлений. У таких людей вирус больше не подавляется в должной степени, что отражается небезопасностью заболевания для них и окружающих [2] [3].
Инфекция ВИЧ-1 с МЛУ — та, которую не удается контролировать никакими из доступных комбинаций АРТ-препаратов, подвергает пациентов повышенному риску госпитализации, прогрессирования заболевания до синдрома приобретенного иммунодефицита (СПИД) и смертельного исхода [4].
Gilead Sciences создала абсолютную защиту от заражения ВИЧ двумя инъекциями в год. Ленакапавир: самый полный обзор в мире.
КАК ЭТО РАБОТАЕТ
В состав каждого вириона ВИЧ-1 входит капсид — белковая оболочка, которая защищает две копии вирусной одноцепочечной РНК и ферментные белки. Жизненный цикл вируса зависит от функционирования капсида на этапах репликации, таких как опосредованный капсидом захват провирусной ДНК ядром клетки, сборка и высвобождение вируса, формирование капсидного ядра.
Ленакапавир (lenacapavir, GS-6207) — первый представитель нового класса антиретровирусных препаратов, называемых ингибиторами капсида ВИЧ.
Ленакапавир представляет собой селективный ингибитор капсидной функции ВИЧ-1. Ленакапавир, связываясь с межмолекулярным интерфейсом между N-концевым доменом одной субъединицы капсидного белка (p24) и C-концевым доменом соседней субъединицы в пределах одного капсидного гексамера, подавляет тройку указанных выше функций следующим образом: путем блокирования связывания белков ядерного импорта с капсидом, путем нарушения функционирования вирусных структурных полипротеинов Gag и Gag–Pol и снижения производства субъединиц капсидного белка, путем нарушения скорости объединения капсидных субъединиц, что приводит к деформации капсида [1] [2] [3] [4] [5] [6].
Итогом применения ленакапавира становится сдерживание репликации вируса.
Ленакапавир обладает противовирусной активностью, специфичной для ВИЧ-1 и ВИЧ-2; в отношении последнего активность снижена в 15–25 раз. На клеточных культурах активность ленакапавира подтверждена против всех групп ВИЧ-1 (M, N, O), включая подтипы A, A1, AE, AG, B, BF, C, D, E, F и G, — со значениями полумаксимальной эффективной концентрации (EC50) от 20 до 160 пмоль.
In vitro ленакапавир характеризуется полной противовирусной активностью против мутантных штаммов ВИЧ-1, устойчивым к АРТ-препаратам четырех основных классов, включая нуклеозидные или нуклеотидные ингибиторы обратной транскриптазы (NRTI, NtRTI), ненуклеозидные ингибиторы обратной транскриптазы (NNRTI), ингибиторы переноса цепи интегразой (INSTI), ингибиторы протеазы (PI) [1] [7] [8].
Ленакапавир синергически (дополняя и усиливая) взаимодействует с указанными классами АРТ-препаратов и не обладает перекрестной резистентностью к другим АРТ-лекарствам, включая ингибиторы созревания.
Благодаря пикомолярной активности ленакапавира, низкому клиренсу и медленной кинетике высвобождения одной подкожной инъекции препарата достаточно для сохранения его должной противовирусной активности на протяжении 6 месяцев. Это же справедливо в случае применения пероральной рецептуры ленакапавира один раз в неделю [1] [9] [10].
Молекулярный дизайн ленакапавира оказался сложнейшей задачей, поскольку итоговое соединение должно было эффективно и селективно воздействовать на капсидные функции ВИЧ, уметь противостоять метаболизирующим лекарственные препараты печеночным ферментам, располагать низким клиренсом и высокой потентностью. Структура соединения и его физико-химические свойства, включая 10 атомов фтора, высокую липофильность и низкую водную растворимость, получились весьма нетипичными, для того чтобы считаться характерными для «лекарственноподобного агента» [11].
КЛИНИЧЕСКИЕ ПОДРОБНОСТИ
Продолжающееся клиническое исследование CAPELLA (NCT04150068) фазы II/III (рандомизированное, двойное слепое, плацебо-контролируемое, многоцентровое, международное) пригласило взрослых пациентов (n=72) с инфекцией ВИЧ-1 и вирусной нагрузкой РНК ВИЧ-1 ≥ 400 копий/мл, указывающей на провал той схемы АРТ, которой они придерживаются.
Основным критерием пригодности включения в испытание была множественная лекарственная устойчивость ВИЧ-1, то есть:
Наличие резистентности как минимум к двум АРТ-препаратам, относящимся по крайней мере к трем классам из четырех основных (NRTI/NtRTI, NNRTI, INSTI, PI). При этом резистентность к эмтрицитабину (emtricitabine; FTC) или ламивудину (lamivudine; 3TC), связанная с наличием мутации M184V/I RT, не использовалась для определения пригодности.
Должно оставаться не более двух полностью активных АРТ-препарата из четырех основных классов, которые могут быть эффективно скомбинированы в подходящую для конкретного испытуемого схему терапии, учитывающую резистентность, переносимость, противопоказания, безопасность, доступность или приемлемость. Из этой схемы исключались: атазанвир (atazanvir; ATV) + кобицистат (cobicistat; COBI), атазанвир + ритонавир (ritonavir; r), эфавиренз (efavirenz; EFV), этравирин (etravirine;ETV) или невирапин (nevirapine, NVP) — ввиду их потенциального взаимодействия с ленакапавиром.
Среди основных характеристик заболевания участников:
число T-клеток CD4+ меньше 200 кл/мл: у 64% человек;
резистентность к NRTI (NtRTI), NNRTI, PI или INSTI: у 99%, 97%, 81% и 69%;
резистентность ко всем четырем основным классам АРТ-препаратов: у 46%;
резистентность к ингибиторам входа (ингибиторам слияния) — энфувиртиду (enfuvirtide; ENF), фостемсавиру (fostemsavir; FTR), ибализумабу (ibalizumab; IBA), маравироку (maraviroc; MVC), применяемым для борьбы с МЛУ: у 9%, 31%, 29% и 67%.
Протокол исследования предусматривал разнесение пациентов по двум когортам — в зависимости от того, как изменился уровень вирусной нагрузки РНК ВИЧ-1 в плазме за период 14–30 дней от момента скрининга до начала испытания.
Если вирусная нагрузка снизилась менее чем на 0,5 log10 копий/мл (то есть наблюдалась стабильная виремия ввиду отсутствия ответа на применяемую АРТ-схему) и она составляла 400 копий/мл и более, пациенты отправлялись в когорту 1.
Если вирусная нагрузка снизилась хотя бы на 0,5 log10 копий/мл или была ниже 400 копий/мл, пациенты примыкали к когорте 2.
Участники когорты 1, продолжающие придерживаться провальной АРТ-схемы, получали пероральный ленакапавир или плацебо — на протяжении 14-дневного этапа функциональной монотерапии. Далее они, уже переключившись на оптимизированную фоновую терапии (ОФТ), переходили к 52-недельному этапу поддерживающей терапии: группе ленакапавира назначали подкожный ленакапавир каждые 6 месяцев (после нагрузочной дозы), группе плацебо — пероральный ленакапавир (в 1-й, 2-й и 8-й дни), а затем подкожный ленакапавир каждые 6 месяцев (после нагрузочной дозы).
Оптимизированная фоновая терапия (ОФТ) — набор АРТ-препаратов, которые выступают основой лечения ВИЧ-инфекции и которые подбираются в индивидуальном порядке, чтобы наилучшим образом контролировать вирус, исходя из особенностей конкретного пациента, таких как предыдущий опыт лечения, резистентность, нежелательные явления.
Участники когорты 2, придерживающиеся ОФТ, вначале проходили 14-дневный этап функциональной монотерапии пероральным ленакапавиром, а затем переходили к 52-недельному этапу поддерживающей терапии подкожным ленакапавиром каждые 6 месяцев (после нагрузочной дозы).
К первичной конечной точке эффективности лечения, установленной пропорцией пациентов из когорты 1, которые по завершении 14-дневного этапа функциональной монотерапии продемонстрировали снижение вирусной нагрузки РНК ВИЧ-1 в плазме как минимум на 0,5 log10 копий/мл, вышли 88% пациентов (n=21/24) в группе ленакапавира и 17% (n=2/12) в группе плацебо (p<0,001) [1].
Усредненная вирусная нагрузка снизилась на 2,10 ±0,15 log10 копий/мл — против прибавления на 0,07 ±0,22 log10 копий/мл.
К двум вторичным конечным точкам эффективности лечения, заявленным пропорцией пациентов в когорте 1, которые по прошествии 26 недель этапа поддерживающей терапии показали вирусную нагрузку РНК ВИЧ-1 ниже 50 копий/мл или ниже 200 копий/мл, вышли соответственно 81% (n=29/36) и 89% (n=32/36) испытуемых. Усредненная вирусная нагрузка снизилась на 2,58 ±1,04 log10 копий/мл.
Указанные показатели для участников из когорты 2 были засвидетельствованы для 83% (n=30/36) и 86% (n=31/36) пациентов. Усредненная вирусная нагрузка снизилась на 2,49 ±1,34 log10 копий/мл.
На 26-й неделе этапа поддерживающей терапии ленакапавиром усредненное число T-лимфоцитов CD4+ увеличилось на 75 клеток/мкл и 104 клеток/мкл — соответственно в когорте 1 и когорте 2. В целом пропорция пациентов с числом T-лимфоцитов CD4+ меньше 50 клеток/мкл снизилась с 24% (n=17/72) до 0% (n=0/67).
Резистентность к ленакапавиру развилась у 11% (n=8/72) испытуемых. Устойчивость к лечению была вызвана аминокислотными заменами M66I, M66I + N74D, K70H или Q67H + K70R; все эти мутации произошли в аминокислотных остатках, которые ранее были выявлены в ходе in vitro проверки ленакапавира на резистентность [2].
Несмотря на развившуюся резистентность к ленакапавиру, половина пациентов (n=4/8) всё же продемонстрировала должную вирусную супрессию (уровень РНК ВИЧ-1 ниже 50 копий/мл) в ходе назначения этого препарата.
Ленакапавир характеризовался приемлемой переносимостью. Каких-либо серьезных нежелательных явлений в ответ на его применение не выявлено.
По истечении 52 недель назначения ленакапавира 83% (n=30/36) и 86% (n=31/36) пациентов в когорте 1 засвидетельствовали вирусную нагрузку РНК ВИЧ-1 соответственно ниже 50 копий/мл и ниже 200 копий/мл [3].
В целом у 13% (n=9/72) испытуемых развилась устойчивость к ленакапавиру, однако у почти половины резистентных участников (n=4/9) была отмечена должная вирусная супрессия.
После 2 лет терапии ленакапавиром статус вирусной супрессии был справедлив для 82% (n=44/54) участников. Усредненная прибавка числа T-лимфоцитов CD4+ за это время составила 122 клетки/мкл. Пропорция пациентов с числом T-лимфоцитов CD4+ меньше 200 клеток/мкл и 50 клеток/мкл снизилась с 64% до 29% и с 24% до 0% соответственно [4].
Частота резистентности к ленакапавиру выросла до 19% (n=14/72). Во всех случаях пациенты имели высокий риск формирования лекарственной устойчивости ввиду либо не полностью активных АРТ-препаратов в рамках ОФТ (n=4), либо неоптимальной приверженности ОФТ (n=10). Половина испытуемых, продолжая придерживаться терапии ленакапавиром, впоследствии достигла должной вирусной супрессии.
КОНТРАРГУМЕНТЫ
Ленакапавир, как и в случае каждого инновационного лекарственного препарата, надежно защищенного патентами, имеет проблемы с ценой и доступностью.
Для американских пациентов, медицинское обслуживание которых не покрыто страховкой, первый год лечения препаратом «Санленка» обойдется в 42 тыс. долларов (без учета скидок и дисконтов) [1].
Согласно оценочному анализу, ленакапавир вполне можно реализовывать по цене 35–40 долларов в год, учитывая себестоимость ингредиентов, производственные расходы и 30-процентную прибыль, — при условии, что препаратом будут пользоваться не менее чем 10 млн человек в год [2]. Столь широкий охват реалистичен, поскольку ленакапавир, назначаемый двумя инъекциями в год, продемонстрировал 100-процентную эффективность в задаче доконтактной профилактики (PrEP) инфекции ВИЧ.
ЧТО ДАЛЬШЕ
«Гилеад» продолжает масштабную клиническую проверку ленакапавира, надеясь найти для него куда более широкое применение в АРТ-схемах ВИЧ-инфекции.
Если говорить о людях, живущих с ВИЧ, которые благодаря стабильному курсу АРТ достигли статуса вирусологической супрессии (уровень РНК ВИЧ-1 < 50 копий/мл или неопределяемый), то ленакапавир изучается в следующих клинических исследованиях.
ARTISTRY-1 (NCT05502341) фазы II/III (n=671) проверяет оправданность перехода с комплексной АРТ-схемы на ежедневную пероральную комбинацию, которая составлена из 75-мг биктегравира (bictegravir; BIC) и 25- или 50-мг ленакапавира (lenacapavir; LEN). Под комплексной АРТ-схемой понимается та терапия, которая была выбрана ввиду резистентности, непереносимости или противопоказаний к однотаблеточным АРТ-схемам.
Ленакапавир и ислатравир, назначаемые каждые 7 дней, не хуже, чем «Биктарви», применяемый каждый день.
NCT05729568 фазы II (n=83) оценивает состоятельность замены ежедневной пероральной АРТ-схемы, состоящей из не более чем двух классов АРТ-препаратов, на назначаемое 1 раз в 6 месяцев инъекционное сочетание подкожного 927-мг ленакапавира с внутривенными теропавимабом (teropavimab, GS-5423) и зинлирвимабом (zinlirvimab, GS-2872), нейтрализующими антителами широкого спектра действия (bNAb).
Терапия ВИЧ-инфекции инъекциями каждые шесть месяцев.
Особое место в бизнес-стратегии «Гилеад» занимает клиническая программа PURPOSE, проверяющая гипотезу, что ленакапавир, назначаемый подкожными инъекциями 1 раз в полгода, пригоден для доконтактной профилактики (PrEP) ВИЧ-инфицирования среди диверсифицированной группы людей с повышенным риском заражения. Во всех испытаниях моноприменение ленакапавира сравнивается с ежедневным пероральным PrEP-препаратом «Трувада» (Truvada, эмтрицитабин + тенофовира дизопроксила фумарат; FTC/TDF). Кроме исследования PURPOSE 1 (NCT04994509), которое изучает комбинацию ленакапавира с «Дескови» (Descovy, эмтрицитабин + тенофовира алафенамида фумарат; FTC/TAF) или «Трувадой», сравнивая ее с назначением только последних [1].
PURPOSE 1 (NCT04994509) фазы III (n=5368): подростки женского пола и молодые женщины (16–25 лет), имеющие секс с мужчинами.
PURPOSE 2 (NCT04925752) фазы III (n=3295): цисгендерные мужчины, трансгендерные женщины, трансгендерные мужчины или небинарные люди (16 лет и старше), имеющие секс с мужчинами.
PURPOSE 3 (NCT06101329) фазы II (n=250): женщины (18 лет и старше), имеющие секс с мужчинами.
PURPOSE 4 (NCT06101342) фазы II (n=250): потребители инъекционных наркотических веществ (18 лет и старше).
PURPOSE 5 (NCT06513312) фазы II (n=262): цисгендерные мужчины (18 лет и старше), имеющие секс с мужчинами, трансгендерными женщинами, трансгендерными мужчинами, цисгендерными женщинами или небинарными людьми.
ЭКСПЕРТНЫЕ КОММЕНТАРИИ
В подавляющем большинстве случаев инфекции вируса иммунодефицита человека назначение антиретровирусной терапии предотвращает репликацию вируса, приводит к вирусологической супрессии (ВС), восстанавливает истощенный пул T-клеток CD4+ и иммунную функцию [1] [2] [3].
Однако высокая эффективность АРТ иногда ограничивается непереносимостью лекарственных препаратов и вызываемыми ими нежелательными явлениями, неоптимальной приверженностью терапии, а также лекарственной устойчивостью, вынуждающей переходить к другим АРТ-схемам.
Лица с высоким уровнем резистентности составляют очень небольшую часть людей, живущих с ВИЧ: согласно оценкам, осуществленным в США в 2012–2017 гг., их не более 1% [4].
Данная популяция пациентов определяется по-разному в разных источниках: например, при резистентности к ≥ 2 классам АРТ-препаратов, при провале или прохождении ≥ 3 линий АРТ, при «большом опыте лечения», при наличии МЛУ и т. п. [5] [6] [7] [8] [9] [10] [11] [12]. В любом случае речь идет о тех людях, живущих с ВИЧ, которые, пробуя несколько АРТ-схем, столкнулись с вирусологической неудачей ввиду либо развившейся резистентности к лекарствам из нескольких АРТ-классов, либо непереносимости, либо проблем с безопасностью [13] [14].
Для того чтобы такие пациенты всё же смогли достичь статуса ВС и затем поддерживать его, составляются схемы оптимизированной фоновой терапии (ОФТ), подбираемые на индивидуальной основе с учетом профиля прежнего лечения, переносимости препаратов, приверженности терапии (комплаентности), лекарственного взаимодействия, доступности препаратов [15] [16] [17]. Ввиду сложностей с подбором конкретных комбинаций АРТ-препаратов данная задача требует выделения дорогостоящих медицинских ресурсов [10]. Однако они необходимы, чтобы избежать риска передачи ВИЧ-инфекции с МЛУ другим людям [18] [19] [20] [21].
«Санленка» (Sunlenca, ленакапавир; LEN), ингибитор капсида ВИЧ, стал третьим по счету препаратом, ориентированным на АРТ лиц с высоким уровнем резистентности. Так, в начале марта 2018 года появился «Трогарзо» (Trogarzo, ибализумаб; IBA), а в начале июля 2020-го — «Рукобиа» (Rucobia, фостемсавир; FTR). Оба этих препарата относятся к ингибиторам входа (ингибиторам слияния), которые предотвращают проникновение вируса в клетку. Все указанные лекарства не применяются монотерапевтически: они назначаются только в составе ОФТ.
Ибализумаб поможет ВИЧ-положительным пациентам, которые не отвечают на применение существующей антиретровирусной терапии.
Есть еще ингибиторы входа «Фузеон» (Fuzeon, энфувиртид; ENF) и «Селзентри» / «Целзентри» (Selzentry / Celsentri, маравирок; MVC), но их использование в контексте борьбы с МЛУ более не оправдано ввиду особых требований к началу лечения, недостаточной эффективности или вопросов к безопасности.
Если стоит вопроса выбора между ленакапавиром (lenacapavir), ибализумабом (ibalizumab) или фостемсавиром (fostemsavir), уместно принять информированное решение, отталкиваясь от систематического обзора и метаанализа, который, охватив ключевые показатели эффективности и безопасности, зарегистрированные в клинических испытаниях, осуществил непрямое сравнение этих препаратов. Следует понимать, что данное исследование профинансировано «Гилеад».
Выяснилось, что на 24–28-й неделях лечения при использовании АРТ-схемы LEN + ОФТ вероятность достичь ВС выше, чем при применении АРТ-схем IBA + ОФТ, FTR + ОФТ или только ОФТ: соответственно в 8,9 раза (95% ДИ [здесь и далее]: 2,1–38,5), 6,6 раза (1,3–32,3) и 12,7 раза (1,7–95,4). При этом рост количества T-клеток CD4+ приблизительно одинаков вне зависимости от выбора из тройки этих препаратов в составе ОФТ [22].
Согласно консенсусным рекомендациям по применению новых АРТ-препаратов против ВИЧ-инфекции с МЛУ, выработанных специалистами Американской академией ВИЧ-медицины и Американским колледжем клинической фармации и опубликованных в начале июня 2024 года, выбор между ленакапавиром, ибализумабом или фостемсавиром в целом равнозначен, но необходимо ответить на вопросы, касающиеся доступности этих лекарственных средств, лекарственного взаимодействия, комплаентности [23].
Согласно анализу выборки из реестра PRESTIGIO (NCT04098315), собирающего данные о ВИЧ-инфицированных с резистентностью ко всем четырем классам АРТ-препаратов, после режимов лечения, включавших FTR, IBA или LEN и с медианой продолжительности 18,7 (5,6–82,7), 14,1 (5,6–39,1) и 30,3 (18,7–33,5) месяца, вирусологическая эффективность (уровень РНК ВИЧ-1 < 200 копий/мл) была справедливой в 67% (n=18/27), 64% (n=7/11) и 90% (n=9/10) случаев. Некоторые пациенты следовали схеме, комбинирующей указанные АРТ-препараты против ВИЧ-1 с МЛУ, в том числе IBA + LEN, IBA + FTR, IBA + FTR + LEN — все с ОФТ [24] [25].
Стандарт первоочередного лечения местнораспространенного или метастатического уротелиального рака (читай, рака мочевого пузыря) ждут масштабные изменения.
Новый фармакологический подход стал первой альтернативой платиносодержащей химиотерапии.
Комбинация препаратов «Падцев» (Padcev, энфортумаб ведотин) и «Китруда» (Keytruda, пембролизумаб) обеспечила лучшие на сегодня клинические исходы.
Это открывает решительно новые перспективы в лечении уротелиальной карциномы: можно надеяться на продление общей выживаемости за пределы двух лет.
За превосходную эффективность приходится расплачиваться побочными реакциями, местами тяжелыми и изредка фатальными.
ЧТО ПРОИЗОШЛО
Сочетание лекарственных препаратов «Падцев» (Padcev, энфортумаб ведотин) и «Китруда» (Keytruda, пембролизумаб) можно использовать в первоочередном лечении местнораспространенного или метастатического уротелиального рака у взрослых пациентов.
Разрешение выдано Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) в середине декабря 2023 года [1].
Европейское агентство по лекарственным средствам (EMA) одобрило «Падцев» с «Китрудой» для лечения местнораспространенного или метастатического уротелиального рака у взрослых пациентов, уже прошедших платиносодержащую химиотерапию и терапию блокатором PD-(L)1 [2].
В России энфортумаб ведотин (enfortumab vedotin) получил регистрацию в конце июня 2023 года под названием «Падцев Онко» при таких же, как в Европе, показаниях [3].
ПОЧЕМУ ЭТО ВАЖНО
Уротелиальный рак, или уротелиальная (переходно-клеточная) карцинома, — тип онкологии, который затрагивает мочевыделительную систему. В 90% случаев речь идет о раке мочевого пузыря, остальные приходятся на почечную лоханку (8%), мочеточник и уретру (2%).
Нынешнее стандартное первоочередное лечение по данному показанию осуществляется химиотерапией, облучением, PD-(L)1-блокаторами «Тецентриком» (Tecentriq, атезолизумаб) или «Китрудой» (Keytruda, пембролизумаб).
Приблизительно половина пациентов не подходит для цисплатин-содержащей химиотерапии ввиду таких причин, как общее состояние здоровья, почечная недостаточность, периферическая нейропатия, нарушение слуха, сердечная недостаточность [1] [2] [3] [4] [5] [6]. Химиотерапия с карбоплатином характеризуется ограниченной активностью и плохо переносится [7] [8].
Применение ингибиторов PD-(L1) ограничено больными, которым нельзя пройти химиотерапию.
Вне зависимости от методов первоочередного лечения местнораспространенного или метастатического уротелиального рака, никакое из них обычно не приводит к существенным положительным исходам, вынуждая переходить к следующей линии терапии.
NB!
Инструкция по медицинскому применения лекарственного препарата «Падцев» (Padcev, энфортумаб ведотин) снабжена «чернорамочным» предупреждением о рисках развития тяжелых и фатальных кожных нежелательных реакций, включая синдром Стивенса — Джонсона (SJS) и токсический эпидермальный некролиз (TEN).
РАНЕЕ
До этого, в начале апреля 2023 года, американский регулятор дозволил применение комбинации «Падцева» (Padcev, энфортумаб ведотин) с «Китрудой» (Keytruda, пембролизумаб) для первоочередного лечения местнораспространенного или метастатического уротелиального рака у взрослых пациентов, которые не пригодны для прохождения химиотерапии с применением цисплатина. Разрешение было выдано условно, то есть лечению предстояло подтвердить свою эффективность [1].
Прежде «Падцев» был разрешен в терапии второй и третьей линии местнораспространенного или метастатического уротелиального рака, где показал весьма примечательную эффективность.
КАК ЭТО РАБОТАЕТ
Энфортумаб ведотин (enfortumab vedotin, ASG-22ME), за разработкой которого стоят «Сиджин» (Seagen), которую купила «Пфайзер» (Pfizer), и «Астеллас фарма» (Astellas Pharma), представляет собой конъюгат моноклонального антитела против нектина-4, несущий цитотоксический монометилауристатин E (MMAE).
Энфортумаб ведотин избирательно связывается с нектином-4 — иммуноглобулиноподобной молекулой клеточной адгезии и опухолеассоциированным антигеном, также известным как белок 4, связанный с рецептором полиовируса (PVRL4, PRR4), который сверхэкспрессирует на поверхности злокачественных клеток при различных солидных опухолях, включая рак мочевого пузыря (в 97% случаев), молочной железы, легких, поджелудочной железы. После интернализации энфортумаба ведотина и протеолитического расщепления линкера происходит связывание MMAE с тубулином, что ингибирует полимеризацию последнего. Это приводит к остановке фазы G2/M клеточного цикла и индуцированию апоптоза опухолевых клеток, сверхэкспрессирующих нектин-4 [1] [2] [3] [4] [5].
Пембролизумаб (pembrolizumab), продвигаемый «Мерк и Ко» (Merck & Co.), является блокатором PD-1.
ЧТО ВЫЯСНИЛОСЬ
Регуляторное одобрение сочетания «Падцева» (Padcev, энфортумаб ведотин) и «Китруды» (Keytruda, пембролизумаб) отталкивалось от результатов двух клинических испытаний.
EV-103/KEYNOTE-869
Продолжающееся клиническое исследование EV-103/KEYNOTE-869 (NCT03288545) фазы Ib/II (рандомизированное, открытое, многоцентровое, международное) пригласило (в когорте K) взрослых пациентов (n=149) с неоперабельным местнораспространенным или метастатическим уротелиальным раком, не подходящих к назначению цисплатин-содержащей химиотерапии и прежде не проходивших лечение.
Участники получали «Падцев» (Padcev, энфортумаб ведотин) в комбинации с «Китрудой» (Keytruda, пембролизумаб) или только «Падцев».
Комбинированная терапия энфортумабом ведотином (enfortumab vedotin) и пембролизумабом (pembrolizumab) обеспечила частоту объективного ответа (ORR) на уровне 65% (95% ДИ [здесь и далее]: 53–75), включая 11% полных ответов (CR) и 54% частичных ответов (PR) [1].
Медиана длительности ответа (DoR) пока не достигнута (10,3 месяца — NR).
Медиана общей выживаемости (OS) составила 22,3 месяца (19,1–NR).
Медиана выживаемости без прогрессирования (PFS) еще не зафиксирована (8,3 месяца — NR).
Лечение только энфортумабом ведотином показало себя менее эффективно:
ORR 45% (34–57), в том числе CR 4% и PR 41%;
медиана DoR 13,2 месяца (6,1–16,0);
медиана OS 21,7 месяца (15,2–NR);
медиана PFS 8,0 месяца (6,1–10,4).
Ранее в этом клиническом исследовании (в когорте A) была засвидетельствована следующая впечатляющая результативность первоочередной комбинации «Падцева» с «Китрудой» [2] [3]:
ORR 74% (58–85), включая CR 16% и PR 58%;
медиана DoR 25,6 месяца (8,3–NR);
24-месячная DoR у 54% пациентов (27–74);
медиана PFS 12,3 месяца (8,0–NR);
медиана OS 26,1 месяца (15,7–NR);
24-месячная OS у 56% пациентов (40–70).
EV-302/KEYNOTE-A39
Продолжающееся опорное клиническое исследование EV-302/KEYNOTE-A39 (NCT04223856) фазы III (рандомизированное, открытое, с активным контролем, многоцентровое, международное), призванное окончательно подтвердить терапевтическую состоятельность первоочередного лечения неоперабельного местнораспространенного или метастатического уротелиального рака сочетанием «Падцева» (Padcev, энфортумаб ведотин) с «Китрудой» (Keytruda, пембролизумаб), осуществляет сравнение этой комбинации со стандартной химиотерапией.
Лекарственный коктейль изучался среди прежде нелеченных пациентов (n=886), пригодных для прохождения цисплатин- или карбоплатин-содержащей химиотерапией, причем вне зависимости от статуса опухолевой экспрессии PD-L1.
Среди основных требований к участникам: нельзя было проходить какую-либо системную терапию местнораспространенного или метастатического уротелиального рака за исключением следующих двух случаев — либо неоадъювантная химиотерапия, либо адъювантная химиотерапия с последующей радикальной цистэктомией, вслед за которыми произошел рецидив заболевания, причем по прошествии не менее чем 12 месяцев после указанного лечения.
Среди основных характеристик испытуемых: медиана возраста 69 лет (22–91), 77% мужчин; у 95% метастатический уротелиальный рак с висцеральным (72%) и печеночным (22%) метастазированием, у 5% местнораспространенный уротелиальный рак; у 85% уротелиальная карцинома, причем у 6% смешанная плоскоклеточная дифференцировка; 46% не пригодны к назначению цисплатина.
Пациенты получали либо энфортумаб ведотин (enfortumab vedotin) с пембролизумабом (pembrolizumab), либо гемцитабин (gemcitabine) с цисплатином (cisplatin) или карбоплатином (carboplatin) — до момента прогрессирования заболевания или неприемлемой токсичности.
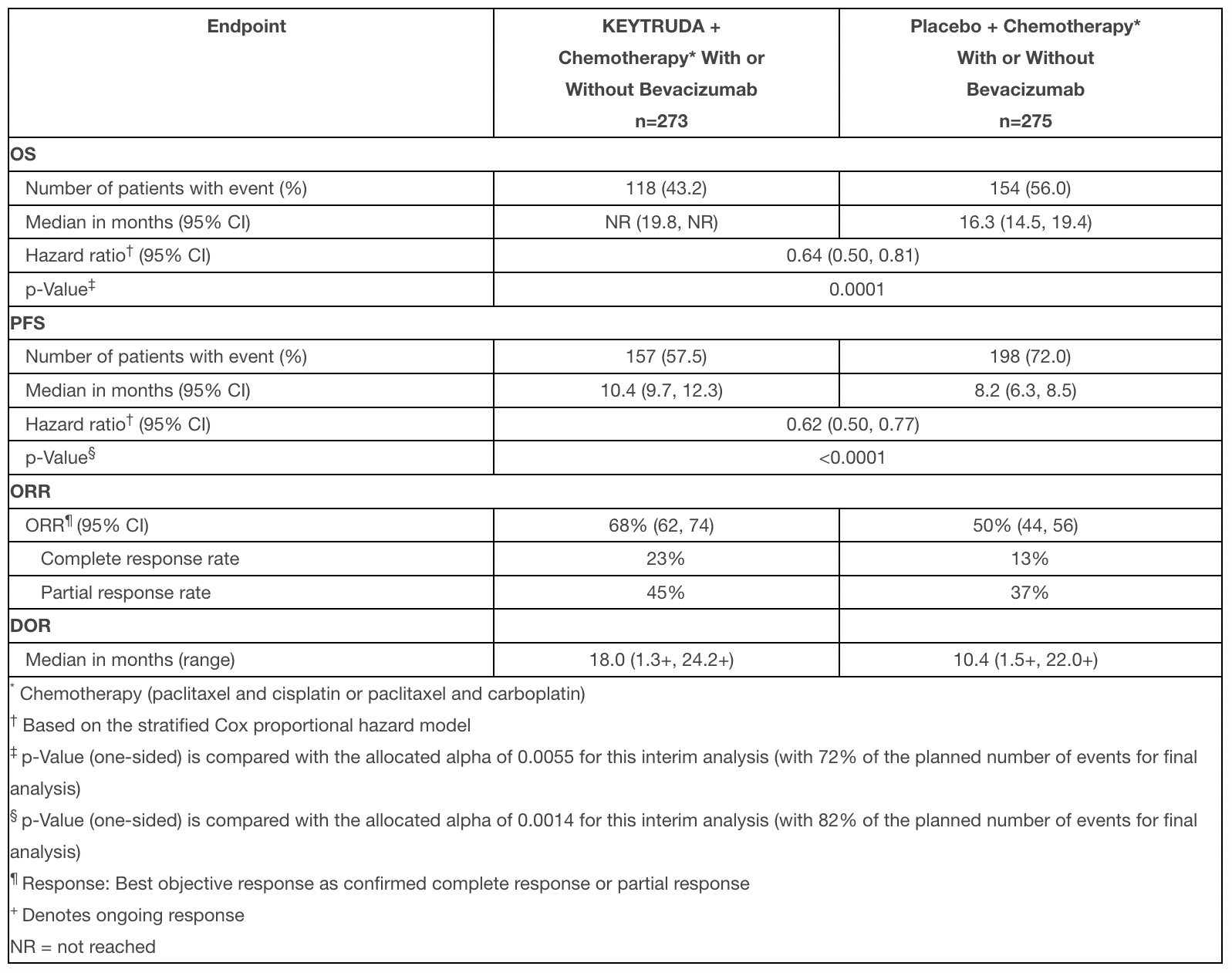
По прошествии наблюдений в течение медианных 17,2 месяца назначение «Падцева» с «Китрудой» вывело медиану общей выживаемости (OS) к 31,5 месяца (95% ДИ [здесь и далее]: 24,5–NR) — против 16,1 месяца (13,9–18,3) в группе стандартной химиотерапии [1] [2].
Применение «Падцева» с «Китрудой» снизило риск смерти на относительных 53%: отношение риска (hazard ratio, HR) 0,47 (0,38–0,58; p<0,00001).
Продление OS и PFS оказалось непротиворечивым среди всех предопределенных подгрупп пациентов, включая разбитых по статусу пригодности к терапии цисплатином и по уровню опухолевой экспрессии PD-L1.
Медиана выживаемости без прогрессирования (PFS) составила 12,5 месяца (10,4–16,6) — против 6,3 месяца (6,2–6,5). Относительный риск прогрессирования заболевания или смерти снизился на 55%: HR 0,45 (0,38–0,54; p<0,00001).
Частота подтвержденного объективного ответа (cORR) получилась равной 68% (63–72), в том числе 29% полных ответов (CR) и 39% частичных ответов (PR), — против cORR 44% (40–49; p<0,00001), включая CR 13% и PR 32%.
Медиана длительности ответа (DoR) в группе «Падцева» с «Китрудой» достигнута не была — против 7 месяцев (6,2–10,2; p<0,00001).
Энфортумаб ведотин с пембролизумабом отсрочил переход к следующей линии терапии: к моменту анализа данных, очередной курс лечения начали проходить 29% пациентов — против 66% в группе химиотерапии.
Безопасность сочетания энфортумаба ведотина с пембролизумабом нельзя назвать приемлемой. Так, из-за нежелательных явлений (НЯ) лечение пришлось временно прервать 73% пациентов, снизить дозу — 42%, полностью прекратить терапию — 35%.
С серьезными НЯ столкнулись 50% испытуемых, с фатальными (острая дыхательная недостаточность, пневмония, пневмонит, интерстициальная болезнь легких) — 4%.
Среди наиболее распространенных НЯ, помимо существенного изменения лабораторных показателей: сыпь (у 68% пациентов), периферическая нейропатия (67%), усталость (51%), зуд (41%), диарея (38%), алопеция (35%), снижение аппетита (33%), снижение веса (33%), тошнота (26%), запор (26%), сухость глаз (24%), инфекции мочевыводящих путей (21%).
ЧТО ДАЛЬШЕ
Комбинация энфортумаба ведотина (enfortumab vedotin) с пембролизумабом (pembrolizumab) проходит два клинических испытания EV-303/KEYNOTE-905 (NCT03924895) и EV-304/KEYNOTE-B15 (NCT04700124) фазы III первоочередного лечения неметастатического мышечно-инвазивного рака мочевого пузыря. Лекарственный коктейль назначается в периоперативный период — до и после оперативного вмешательства, предусматривающего радикальную цистэктомию с тазовой лимфаденэктомией. В первом исследовании сравнение осуществляется с применением только хирургии, во втором — хирургии с химиотерапией.
Энфортумаб ведотин также изучается в лечении широкого спектра солидных опухолей. Клиническое исследование EV-202 (NCT04225117) фазы II пригласило пациентов с местнораспространенными или метастатическими онкологическими заболеваниями, такими как рак молочной железы (HR+/HER2− или трижды негативный), немелкоклеточный рак легкого (НМРЛ), рак головы и шеи, рак желудка, пищевода или пищеводно-желудочного перехода.
В клиническом исследовании EV-104 (NCT05014139) фазы I тестируется интравезикулярная рецептура энфортумаба ведотина для лечения мышечно-инвазивного рака мочевого пузыря после провала иммунотерапии бациллой Кальметта — Герена (БЦЖ).
БИЗНЕС
Согласно прогнозам «Астеллас фарма» (Astellas Pharma), общемировые годовые продажи «Падцева» (Padcev, энфортумаб ведотин) могут добраться до 400–500 млрд иен (2,6–3,2 млрд долларов).
В 2023 году «Падцев», перешагнувший планку в 1 млрд долларов реализации, стал бестселлером: препарат принес 1,1 млрд долларов.
ЭКСПЕРТНЫЕ КОММЕНТАРИИ
Энфортумаб ведотин (enfortumab vedotin) и пембролизумаб (pembrolizumab) — эффективные лекарственные препараты для монотерапии местнораспространенного или метастатического уротелиального рака [1] [2] [3] [4].
Согласно доклиническим исследованиям несущих цитотоксическую нагрузку конъюгатов моноклональных антител, включая энфортумаб ведотин, такие препараты характеризуются признаками иммуногенной гибели клеток, включая высвобождение ассоциированых с повреждением молекулярных паттернов (DAMP) [5] [6] [7] [8]. Последние распознаются клетками врожденного и адаптивного иммунитета, что в конечном итоге приводит к захвату опухолевых клеток антигенпрезентирующими клетками (APC) с последующей перекрестной презентацией опухолевых антигенов цитотоксическим T-лимфоцитам (CTL). Они индуцируют антиген-специфический ответ, который дополнительно усиливается блокаторами PD-(L)1 вроде пембролизумаба.
Вопрос с необходимостью достижения высокой эффективности у первоочередного фармакологического лечения уротелиального рака стоит весьма остро. Во-первых, многие пациенты вообще не приступают к первой линии терапии, в том числе из-за ее недостаточной эффективности и плохой переносимости [9] [10].
Во-вторых меньше половины пациентов переходят к последующим линиям терапии [11] [12], то есть первая линия должна обеспечивать максимальный контроль над заболеванием.
В-третьих, половина пациентов не пригодна к первоочередной цисплатин-содержащей химиотерапии [13] [14] [15], и выживаемость таких больных низкая, что обусловлено сопутствующими заболеваниями и недостаточной активностью альтернативного карбоплатина.
В-четвертых, меньшая пропорция пациентов, которые прошли первоочередной химиотерапевтический курс с карбоплатином, могут воспользоваться второлинейными блокаторами PD-(L)1: по причине более низкой частоты ответов и меньшей продолжительности жизни по сравнению с применением цисплатина [16] [17].
Всё идет к тому, что комбинация энфортумаба ведотина и пембролизумаба станет новым стандартом первой линии терапии уротелиального рака, поскольку предоставляемые ею клинические исходы (показатели ответа и его длительность, частота контроля заболевания, общая выживаемость) являются самыми лучшими на сегодня.
Для сравнения: сочетание PD-1-блокатора «Опдиво» (Opdivo, ниволумаб) авторства «Бристол-Майерс Сквибб» (Bristol-Myers Squibb) с гемцитабином и цисплатином, проверенное в клиническом испытании CheckMate 901 (NCT03036098) фазы III, вывело общую выживаемость (OS) на медианный уровень 21,7 месяца (95% ДИ [здесь и далее]: 18,6–26,4) — против 18,9 месяца (14,7–22,4) в группе только химиотерапии, тем самым снизив относительный риск смерти на 22%: отношение риска (hazard ratio, HR) 0,78 (0,63–0,96; p=0,02). Частота общего ответа (ORR) составила 58%, включая 22% полных ответов (CR), — против ORR 43% и CR 12% [18].
«Модерна» (Moderna) в сотрудничестве с «Мерк и Ко» (Merck & Co.) разработала mRNA-4157 (V940) — персонализированную противораковую вакцину, которая производится отдельно под каждого пациента с четким прицелом на специфику его заболевания.
На данном этапе собраны весьма обнадеживающие клинические результаты лечения прошедшей хирургическую резекцию меланомы, превосходящие стандартную терапию.
Продемонстрированы примечательные исходы лечения неоперабельного рака головы и шеи. Продолжается клиническая проверка терапии рака легкого, рака почки, рака мочевого пузыря, немеланомного рака кожи.
Механистическое обоснование концепции персонализированных противораковых вакцин выглядит разумным и состоятельным. Мол, они запускают доселе невиданный мощный противоопухолевый иммунный ответ, причем высокоспецифичный, учитывающий особенности онкопатологии каждого конкретного пациента. Но пока шквал потенциально благотворных T-клеточных реакций не превратится в устойчивую и продолжительную клиническую ремиссию, будущего у противораковых вакцин попросту нет. И здесь обломали зубы десятки и сотни разработчиков. Но у «Модерна», кажется, получилось.
Если всё и дальше будет идти по плану, индивидуализированная неоантигенная мРНК-онковакцина mRNA-4157 (V940) получит ускоренное регуляторное одобрение в 2025 году.
Относительно несложный процесс выпуска и применения mRNA-4157 (V940) идет вразрез с комплексным и дорогостоящим производственным циклом и назначением терапии T-клетками с химерным антигенным рецептором (CAR), препараты на базе которой стали настоящим прорывом в лечении запущенных онкогематологических заболеваний.
Тем временем явных успехов добилась немецкая «Байонтек» (BioNTech). Персонализированная противораковая мРНК-вакцина аутоген цевумеран (autogene cevumeran), изучаемая совместно с «Дженентек» (Genentech) в составе «Рош» (Roche), весьма достойно показала себя в лечении рака поджелудочной железы.
Персонализированная иммунотерапия обеспечила уверенное продление жизни для трети пациентов с протоковой аденокарциномой поджелудочной железы.
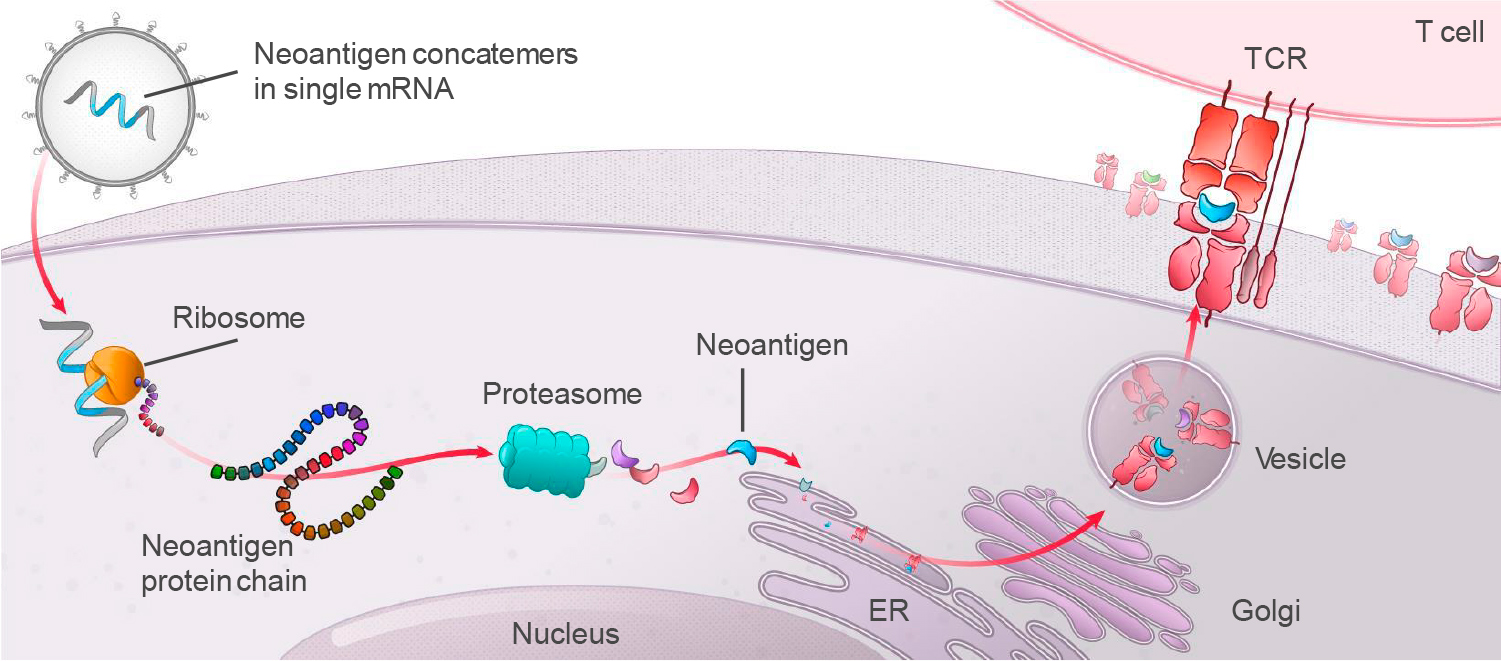
МЕХАНИЗМ ДЕЙСТВИЯ
mRNA-4157 (V940) представляет собой персонализированную противораковую вакцину, которая, следуя концепции прецизионной медицины, должна резко улучшить эффективность лечения онкологических заболеваний.
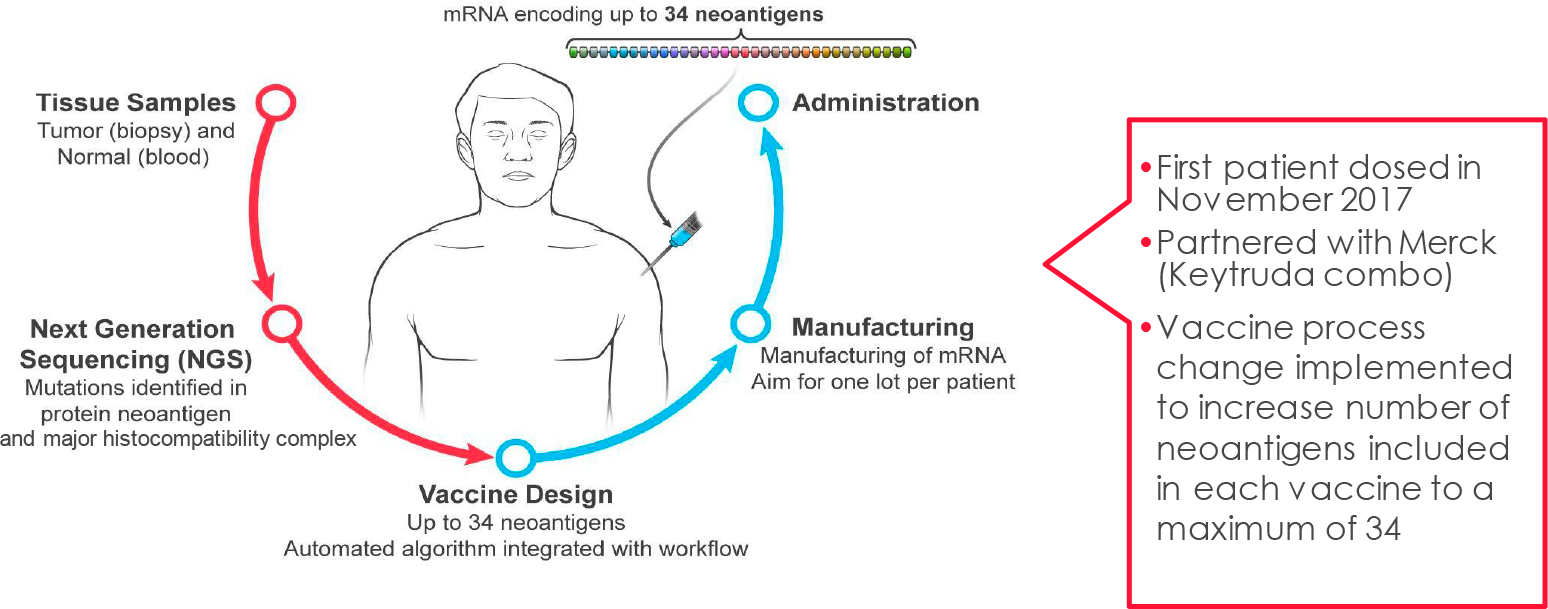
Из опухолей и образцов крови пациента выделяют только ему свойственные неоантигены — антигены, закодированные мутантными генами, специфичными для раковых клеток в данных новообразованиях (последние развиваясь и прогрессируя, мутируют в обязательном порядке). При этом в состав онковакцины входят равно как эпитопы неоантигенов, которые были обнаружены в ходе ex vivo экспериментов над иммунными клетками пациента, так и эпитопы неоантигенов из всего экзома, которые, согласно прогностическому биоинформационному алгоритму, являются иммуногенными, то есть способными запустить в организме благотворные иммуностимулирующие реакции.
In silico собирается мРНК-последовательность, кодирующая одновременно до 34 эпитопов (неоантигенный конкатемер). Далее мРНК-молекулы инкапсулируются в фирменную оболочку из липидных наночастиц (LPN), которая наделяет готовый онковакцинный препарат толерантностью, минимизирует токсичность при многократном введении, помогает укрыться от надзора иммунной системы и защититься от ферментативного распада.
После внутримышечного введения персонализированной противораковой вакцины в организм антигенпрезентирующие клетки (APC) захватывают и транслируют мРНК-инструкции с дальнейшей экспрессией соответствующих эпитопов на своей поверхности. Итогом становится индуцирование специфических иммунных ответов со стороны цитотоксических T-клеток CD8+ и T-клеток памяти CD4+.
Доставка в организм сразу целого множества опухолеспецифических антигенов (TSA) должна резко повысить вероятность успешных клинических исходов, поскольку иммунная система начинает генерировать многовекторный T-клеточный ответ на неоантигенные пептиды, которые ей были презентированы. Другими словами, иммунная система проходит «обучение», по итогам которого усиливается ее способность распознавать и уничтожать опухолевые клетки.
В терминологии «Модерна» мРНК-вакцина mRNA-4157 (V940) относится к индивидуализированной неоантигенной терапии (Individualized Neoantigen Therapy, INT).
Разработка mRNA-4157 (V940) осуществляется совместно с «Мерк и Ко» (Merck & Co.), с которой «Модерна» в июне 2016 года вошла в стратегическое соглашение: первая выплатила второй 200 млн долларов авансом [1]. В мае 2018 года партнерство было расширено, что повлекло за собой финансовое вливание дополнительных 125 млн долларов [2]. В октябре 2022 года долгосрочный альянс укрепился еще сильнее: «Мерк и Ко» полностью поверила в успех mRNA-4157 (V940) [3].
КЛИНИЧЕСКАЯ ПРОВЕРКА
Продолжающееся клиническое исследование KEYNOTE-603 (NCT03313778) фазы I (нерандомизированное, открытое, многоцентровое) является основополагающим для дальнейшей разработки персонализированной противораковой вакцины mRNA-4157 (V940), поскольку изучает ее применимость различными схемами для лечения широкого ассортимента солидных опухолей.
Исследование охватывает такие диагнозы, как немелкоклеточный рак легкого (НМРЛ), мелкоклеточный рак легкого (МРЛ), меланома, уротелиальная карцинома (рак мочевого пузыря), плоскоклеточная карцинома головы и шеи (не индуцированная вирусом папилломы человека), протоковая аденокарциномы поджелудочной железы, а также любые злокачественные новообразования с высокочастотной микросателлитной нестабильностью (MSI-H) или дефектами в системе репарации ошибочно спаренных оснований ДНК (dMMR): например, колоректальный рак, аденокарцинома желудка и пищевода, рак эндометрия.
Параллельно осуществляется клиническое исследование KEYNOTE-942 (NCT03897881) фазы IIb (рандомизированное, открытое, многоцентровое, международное) среди пациентов с меланомой кожи (высокорисковой, на стадии IIIB–D или IV), которая метастазировала в лимфоузел и затем прошла полную хирургическую резекцию, но всё еще обладает высоким риском рецидива.
На момент рандомизации заболевание участников должно было быть в состоянии ремиссии, без локорегионарного рецидива, отдаленных метастазов, метастазирования в головной мозг.
Поставлена задача выяснить, оправдано ли добавление персонализированной мРНК-онковакцины mRNA-4157 (V940) к «Китруде» (Keytruda, пембролизумаб), блокатору PD-1 авторства «Мерк и Ко» (Merck & Co.), в целях продления безрецидивной выживаемости (RFS), если сравнивать с назначением только пембролизумаба (pembrolizumab). Предпосылки для этого очевидны: в адъювантных условиях (после резекции) опухоль отсутствует, пациент еще не проходил слишком много курсов химиотерапии, а его иммунная система относительно здорова.
Относительно недавно запущены следующие клинические испытания, оценивающие комбинацию mRNA-4157 (V940) и «Китруды» в задаче продления бессобытийной выживаемости после хирургической резекции, то есть удержания пациента в статусе ремиссии:
INTerpath-001 (V940-001, NCT05933577) фазы III. Меланома кожи (на стадии IIB–C, III или IV) после хирургической резекции, без признаков заболевания, с высоким риском рецидива, ранее не проходившая системную терапию. mRNA-4157 (V940) + пембролизумаб — сравнение с пембролизумабом. Конечные точки: RFS, DMFS, OS.
INTerpath-002 (V940-002, NCT06077760) фазы III. Немелкоклеточный рак легкого (НМРЛ) [на стадии II, IIIA или IIIB/N2] после хирургической резекции, без признаков заболевания, прошедший хотя бы одну линию адъювантной платиносодержащей химиотерапии. mRNA-4157 (V940) + пембролизумаб — сравнение с пембролизумабом. Конечные точки: DFS, OS, DMFS, LCSS.
V940-004 (NCT06307431) фазы II. Почечно-клеточная карцинома (рак почки) после хирургической резекции, без признаков заболевания, с промежуточно-высоким или высоким риском рецидива, ранее не проходившая какой-либо лечение. mRNA-4157 (V940) + пембролизумаб — сравнение с пембролизумабом. Конечные точки: DFS, OS, DMFS.
V940-005 (NCT06305767) фазы II. Мышечно-инвазивная уротелиальная карцинома (рак мочевого пузыря) после хирургической резекции, с высоким риском рецидива, ранее не проходившая системную терапию. mRNA-4157 (V940) + пембролизумаб — сравнение с пембролизумабом. Конечные точки: DFS, OS, DMFS.
INTerpath-007 (V940-007, NCT06295809) фазы II/III. Плоскоклеточная карцинома кожи (на стадии II–IV без отдаленных метастазов), местнораспространенная, операбельная, ранее не проходившая какую-либо терапию. mRNA-4157 (V940) + пембролизумаб до и после хирургической резекции — сравнение с хирургической резекцией ± пембролизумаб. Конечные точки: EFS, ORR, FFS rate, pCR rate, mPR rate, DFS, DSS, OS.
Сокращения: DFS, выживаемость без признаков заболевания; DMFS, выживаемость без отдаленного метастазирования; DSS, выживаемость с учетом заболевания; EFS, бессобытийная выживаемость; FFS, свобода от хирургического вмешательства; LCSS, выживаемость, специфическая при раке легкого; mPR, большой патоморфологический ответ; ORR, частота общего ответа; OS, общая выживаемость; pCR, полный патоморфологический ответ; RFS, безрецидивная выживаемость.
Geneos Therapeutics придумала, как вылечить распространенную гепатоцеллюлярную карциному.
ЛЕЧЕНИЕ МЕЛАНОМЫ
По прошествии наблюдений на протяжении медианных 2 лет (23–24 месяца) за пациентами с меланомой из KEYNOTE-942 (NCT03897881) фазы IIb, которые получили либо mRNA-4157 (V940) с «Китрудой» (Keytruda, пембролизумаб), либо только пембролизумаб (pembrolizumab), экспериментальное комбинированное иммунотерапевтическое лечение клинически значимым (но не статистически!) образом улучшило безрецидивную выживаемость (RFS), снизив риск рецидива или смертельного исхода на 44%, если сравнивать с монотерапией пембролизумабом: отношение риска (hazard ratio, HR) 0,56 (95% ДИ [здесь и далее]: 0,31–1,02; двусторонний p=0,053) [1] [2] [3].
[membership level=»2,3″ show_noaccess=»true»]
В статусе RFS на протяжении 12 месяцев оказались 83% и 77% пациентов, 18 месяцев — 79% и 62% [4].
Непротиворечивое продление RFS, обеспеченное персонализированной мРНК-онковакциной с блокатором PD-1, было отмечено вне зависимости от уровня опухолевой мутационной нагрузки (TMB), балла опухолевого воспаления (TIS) и статуса PD-L1. Соответствующие показатели HR таковы [5]:
высокая TMB (175 мутаций на экзом, или ≥ 10 мутаций на мегабазу) и невысокая TMB: 0,65 (0,28–1,49) и 0,59 (0,24–1,42);
высокий TIS и невысокий TIS (отсечение 4,56): 0,58 (0,21–1,59) и 0,53 (0,25–1,10);
опухоли, положительные (балл CPS ≥ 1) и отрицательные по PD-L1: 0,49 (0,23–1,04) и 0,16 (0,04–0,69).
По истечении 18 месяцев наблюдений с отдаленным метастазированием меланомы или смертельным исходом столкнулись 8% пациентов в группе комбинированного лечения и 24% — монотерапии «Китрудой». Частоты выживаемости без отдаленного метастазирования (DMFS) на протяжении 12 месяцев составили 93% и 89%, 18 месяцев — 92% и 77% [6] [7] [3].
Таким образом, добавление индивидуализированной противораковой вакцины mRNA-4157 (V940) к блокатору PD-1 пембролизумабу обеспечило клинически значимое улучшение показателя DMFS, снизив риск развития отдаленных метастазов или смерти на 65% в сравнении с лечением только «Китрудой» (HR 0,35 [0,15–0,83]; односторонний p=0,0063).
Установлено, что если до лечения не обнаруживалась циркулирующая опухолевая ДНК (ctDNA), которая отражает наличие минимально остаточной болезни (MRD) после опухолевой резекции и является биомаркером безрецидивной выживаемости при высокорисковой меланоме, прошедшей хирургическую операцию, то и ответ на терапию был лучше [8].
Так, среди пациентов с исходным негативным ctDNA-статусом показатели RFS и DMFS получились лучше при сравнении комбинированного лечения с монотерапией: риск рецидива или смерти и риск отдаленного метастазирования или смерти снизились на соответствующих 77% (HR 0,23 [0,10–0,53]) и 95% (HR 0,05 [0,01–0,38]). Впрочем, и при позитивном ctDNA-статусе были отмечены тенденции к улучшению RFS и DMFS, обеспеченные сочетанной терапией. Однако небольшая пациентская выборка не позволила сделать окончательные выводы, что эффективность терапевтической персонализированной противораковой вакцины mRNA-4157 (V940) в целом не зависит от изначального статуса ctDNA.
Индивидуализированная неоантигенная терапия mRNA-4157 (V940) работала лучше стандартного лечения вне зависимости от статуса BRAF: мутантный или дикого типа.
После медианных 3 лет наблюдений выяснилось, что лечение меланомы при помощи mRNA-4157 (V940) с пембролизумабом, клинически значимым образом улучшило показатели RFS и DMFS, снизив риск рецидива или смерти на 49% (HR 0,51 [0,29–0,91]; односторонний номинальный p=0,0095) и снизив риск развития отдаленных метастазов или смерти на 62% (HR 0,38 [0,17–0,86]; односторонний номинальный p=0,0077), если сравнивать с назначением только пембролизумаба [9].
[/membership]
СУТЬ
Длительное применение блокаторов PD-1 в адъювантном лечении резецированной высокорисковой меланомы в целях предупреждения ее возвращения — стандартная и эффективная клиническая практика [1] [2] [3], хотя и сопровождающаяся высоким риском хронической токсичности [4]. Однако терапевтическая персонализированная противораковая вакцина mRNA-4157 (V940) уже готова, кажется, обновить эту парадигму.
[membership level=»2,3″ show_noaccess=»true»]
Во всяком случае получены надежные доказательства улучшения RFS и DMFS в сравнении с нынешними терапевтическими подходами.
Так, наблюдения в течение 3,5 лет за пациентами, прошедшими годичный курс пембролизумаба (pembrolizumab), установили, что в статусах RFS и DMFS оставались 60% и 65% человек, а после 5 лет наблюдений — 55% и 61% [5] [6]. В случае годичной терапии ниволумабом (nivolumab) 4-летние показатели RFS и DMFS оказались справедливыми для 52% и 59% пациентов [7].
То есть в долгосрочной перспективе приблизительно половина пациентов сталкиваются с возвращением меланомы (или смертью), что вынуждает переходить к следующим линиям терапии. Таким образом, перед mRNA-4157 (V940) стояла задача как можно более продолжительного по времени сдерживания рецидива (RFS), который может усугубиться метастазированием (DMFS). И мРНК-онковакцина с таким упреждением справилась, хотя, впрочем, пока без подтвержденного статистически значимого расхождения с монотерапией пембролизумабом. Скорее всего, на это повлияла скромная по охвату выборка испытуемых.
Тем временем пембролизумаб засвидетельствовал оправданность своего одновременно назначения в неоадъювантном и адъювантном периодах, то есть равно как до резекции, так и после нее [8]. Сочетание ипилимумаба (ipilimumab) с ниволумабом продемонстрировало собственную пригодность в неоадъюватном периоде при определенных условиях [9] [10]. Гипотеза состоит в том, что еще не удаленная опухоль имеет достаточную массу, чтобы вызвать адекватную активацию и экспансию системного противоопухолевого T-клеточного ответа — такого же по силе, как в случае иммунотерапии рецидивирующей меланомы [11].
Открытым и важным остается вопрос с продлением общей выживаемости (OS): пока нет надежных данных, продлевают ли жизнь блокаторы PD-1, притом что конечные точки RFS и DMFS являются лишь суррогатными для OS, и делать какие-то определяющие заключения на их основе можно лишь с натяжкой [12].
В любом случае «Модерна» надо постараться с развитием mRNA-4157 (V940): в постковидную эпоху больше нет сверхдоходов от продажи вакцин против SARS-CoV-2.
С точки зрения бизнеса «Модерна» поступила чрезвычайно разумно, что потратила длительное время на проведение клинического испытания фазы IIb, прежде чем в конце июля 2023 года запустила решающую фазу III [13], поскольку было слишком много неудачных клинических исследований противораковых вакцин. Не всем нравится подобный промежуточный подход к разработке, но консервативная стратегия оправдана.
Чрезвычайная ситуация, связанная с пандемией коронавирусной инфекции COVID-19, официально прекращена.
[/membership]
ЛЕЧЕНИЕ РАКА ГОЛОВЫ И ШЕИ
Промежуточный анализ клинических исходов KEYNOTE-603 (NCT03313778) фазы I в когорте лечения неоперабельной (метастатической или рецидивирующей) плоскоклеточной карциномы головы и шеи (HNSCC) [ротовой полости, ротоглотки, гортаноглотки, гортани], не индуцированной вирусом папилломы человека (HPV−), ранее не проходившей терапию какими-либо ингибиторами иммунных контрольных точек (ИИКТ), установил следующие результаты [1].
[membership level=»2,3″ show_noaccess=»true»]
На начало октября 2020 года, комбинация из индивидуализированной мРНК-онковакцины mRNA-4157 (V940) и PD-1-блокатора «Китруды» (Keytruda, пембролизумаб) обеспечила частоту общего ответа (ORR) на уровне 50% (n=5/10): полный ответ (CR) зафиксирован у 20% (n=2/10) пациентов, частичный ответ (PR) — у 30% (n=3/10). Стабилизация заболевания (SD) отмечена у 40% (n=4/10). С прогрессированием (PD) столкнулся один человек. Таким образом, частота контроля заболевания (DCR), как суммы CR, PR и SD, составила 90% (n=9/10).
Медиана длительности ответа (DoR) еще не созрела. Медиана выживаемости без прогрессирования (PFS) — 9,8 месяца.
Что примечательно, 4 из 5 респондентов ответили на лечение после двух доз пембролизумаба (pembrolizumab)— еще до назначения mRNA-4157 (V940). После того как они получили вакцину, ответы углубились: к примеру, два пациента с частичной ремиссией перешли к полной. Еще один человек, заболевание которого прогрессировало на фоне «Китруды», после вливания вакцины наконец-то засвидетельствовал частичный ответ.
Для сравнения: монотерапия пембролизумабом рецидивирующей или метастатической плоскоклеточной карциномы головы и шеи выводит ORR и медиану PFS к 14,6% и 2,0 месяца. Более того, экспериментальное вакцинное противораковое лечение превзошло стандартную первоочередную терапию, предполагающую назначение «Китруды» вместе с химиопрепаратами и выдающую ORR 36% и медиану PFS 4,9 месяца [2] [3] [4].
Любопытные выводы сделаны сообразно анализу предиктивных биомаркеров. Отмечена тенденция к благоприятному клиническому ответу у пациентов с опухолями, характеризующимися повышенным уровнем воспаления, о котором свидетельствуют баллы GEP (профиль генной экспрессии) и CYT (цитолитическая активность). При этом, однако, корреляции с опухолевой мутационной нагрузкой (TMB) не выявлено.
Балл GEP, во-первых, отражает уровень РНК-экспрессии 18 воспалительных генов, имеющих отношение к антигенной презентации, экспрессии хемокинов, цитолитической активности и адаптивной иммунорезистентности (включая PD-L1), и, во-вторых, указывает на уровень T-клеточного воспаления в опухолевом микроокружении (TME). Балл CYT рассчитывается на основе транскрипционных уровней двух ключевых цитолитических эффекторов — гранзима A и перфорина.
Дальнейший анализ на начало мая 2023 года с медианой наблюдений в течение 9,0 (2,7–45,5) месяца и охвативший больше пациентов, установил следующие исходы: ORR 27% (11–50), включая CR 9% и PR 18%, DCR 64% (41–83), расчетные медианы PFS 3,5 (2,7–9,0) месяца и общей выживаемости (OS) 25,0 (9,0–NE) месяца [5].
[/membership]
СУТЬ
Пациенты с плоскоклеточной карциномой головы и шеи (HNSCC), отрицательной по вирусу папилломы человека (HPV−), характеризуются плохим прогнозом: пятилетняя частота выживаемости не превышает 50% [1].
[membership level=»2,3″ show_noaccess=»true»]
Считается, что ограниченный рост стойких клинических ответов на терапевтическую блокаду PD-(L)1 обусловлен снижением эффекторной цитолитической активности и клональным разнообразием [2] [3] [4] [5].
Поскольку индивидуализированная мРНК-онковакцина mRNA-4157 (V940), которая кодирует до 34 опухолевых неоантигенов, вызывает специфическую активацию противоопухолевых T-клеток, что было подтверждено успешным лечением меланомы, имело смысл опробовать ее назначение в связке с PD-1-блокатором пембролизумабом (pembrolizumab).
Иммуноонкологический коктейль продемонстрировал признаки активации иммунных реакций, которые обозначили себя среди ответивших на лечение в том числе снижением уровня циркулирующей опухолевой ДНК (ctDNA). «Более теплые» опухоли, такие как в случае HNSCC (HPV−), располагают более благоприятным опухолевым микроокружением (TME) для генерации T-клеточных ответов, индуцированных сочетанием mRNA-4157 (V940) c пембролизумабом. Кроме того, добавление неоантигенов, принесенных в составе персонализированной противораковой вакцины, снижает планку уровня опухолевой мутационной нагрузки (TMB), необходимого для должного терапевтического ответа на пембролизумаб.
Предварительные выводы таковы: при лечении HNSCC (HPV−) добавление мРНК-вакцины к стандартному пембролизумабу продлит общую выживаемость как минимум вдвое, если сравнивать с использованием только последнего.
[/membership]
ПРОТИВОРАКОВЫЕ ВАКЦИНЫ: ХОРОШО, ДА МАЛО
Несмотря на многообещающий потенциал противораковых вакцин, это направление биотехнологий развивается чрезвычайно медленно. Существует ряд препятствующих проблем: гетерогенность опухолевых антигенов и их мутационный разброс, отсутствие унифицированного сигнального пути распознавания антигенов с последующей активацией иммунного ответа, множество способов ухода раковых клеток от иммунологического надзора, выбор нужного иммуностимулирующего адъюванта, оптимальный путь доставки вакцины в организм.
Тем не менее на клиническом конвейере есть немало перспективных противораковых вакцин, включая персонализированные.
Ну а пока что в мире одобрены только две персонализированные противораковые вакцины: «Онкофаг» (Oncophage, витеспен) и «Провендж» (Provenge, сипьюлейсел-T).
Есть еще тройка противораковых препаратов на основе онколитических вирусов — «Онкорин» (Oncorine), «Имлайджик» (Imlygic, талимоген лагерпарепвек) и «Делитакт» (Delytact, тесерпатурев), применяемых в лечении соответственно карциномы головы и шеи, меланомы и злокачественной глиомы, — но к онковакцинам их можно отнести с натяжкой. Впрочем, механизм действия (хотя их несколько) этих лекарственных средств весьма близок: иммунной системе презентируются опухолевые антигены, накапливающиеся ввиду целенаправленного разрушения раковых клеток, индуцированного онколитическими вирусами.
Особняком стоит противотуберкулезная вакцина БЦЖ (бацилла Кальметта — Герена), которая почти полвека с большим успехом применяется в иммунотерапии рака мочевого пузыря без инвазии в мышечный слой (NMBIC).
«Онкофаг», ранее известная как «Профаг» (Prophage), представляет собой комплекс, включающий белок теплового шока (HSP) gp96 и фрагментов пептидов, полученных из опухолевой ткани пациента. Аутологичный антигенный комплекс витеспен (vitespen, HSPPC-96) стимулирует резидентные дендритные клетки, которые активируют цитотоксические T-лимфоциты (CTL) и T-хелперы — ключевые компоненты каскада противоопухолевого иммунного ответа [1].
«Онкофаг» разработана «Антидженикс» (Antigenics), которая в январе 2011 года сменила название на «Адженус» (Agenus) [2].
«Онкофаг» получила регуляторное разрешение только в России, где была одобрена в апреле 2008 года [3] [4].
В ноябре 2009 года «Антидженикс» отозвала заявку на регистрацию «Онкофага» в Европейском союзе, после того как экспертный комитет при Европейском агентстве по лекарственным средствам (EMA) пришел к заключению, что онковакцина не в силах существенно продлить безрецидивную выживаемость. Кроме того, производитель не предоставил полной информации относительно состава «Онкофага» и его производственного процесса, а также механизма действия при почечно-клеточной карциноме и выбора нужной дозы [5].
Применение «Онкофага» снижает риск прогрессирования заболевания или смерти (PFS) на 41% (HR 0,59 [0,37–0,94]; p=0,026) и риск смерти (OS) на 46% (HR 0,54 [0,30–0,97]; p=0,036) [6] [7].
[/membership]
«ПРОВЕНДЖ»
Персонализированная противораковая вакцина «Провендж» (Provenge, сипулейцел-T) предназначена для лечения бессимптомного или минимально симптоматического метастатического кастрационно-резистентного рака предстательной железы.
[membership level=»2,3″ show_noaccess=»true»]
«Провендж» производится из собственных мононуклеарных клеток периферической крови (PBMC) пациента, включая антигенпрезентирующие клетки (APC), T- и B-лимфоциты, естественные киллеры (NK), которые затем активируются ex vivo рекомбинантным человеческим белком PAP-GM-CSF. Последний представляет собой композицию из простатической кислой фосфатазы (PAP) — антигена, высокоэкспрессирующего в опухолевых тканях при раке простаты, и гранулоцитарно-макрофагального колониестимулирующего фактора (GM-CSF) — активатора иммунных клеток. После внутривенного вливания сипулейцела-T (sipuleucel-T, APC-8015) запускаются процессы гуморального и T-клеточного иммунного противоопухолевого ответа [1] [2].
«Провендж» разработана «Дендрион» (Dendreon), которую в феврале 2015 года за 495 млн долларов купила канадская «Валеант фармасьютикалс интернешнл» (Valeant Pharmaceuticals International), уже в июле 2017-го продавшая ее за 820 млн долларов китайскому конгломерату Sanpower Group [3] [4] [5] [6]. Через год последняя перепродала «Дендрион» за 868 млн долларов китайской Nanjing Cenbest, крупному розничному ретейлеру, решившему заняться биотехнологическим бизнесом. Впрочем, сделка по сути номинальная, ведь Sanpower является крупнейшим акционером Nanjing Cenbest [7].
В апреле 2010 года «Провендж» была одобрена Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) [8].
В сентябре 2013 года «Провендж» получила маркетинговое разрешение Европейского агентства по лекарственным средствам (EMA), однако в мае 2015-го регистрация была прекращена по просьбе «Дендрион» ввиду коммерческих соображений [9].
Терапевтическая эффективность «Провендж» весьма скромная: персонализированная противораковая вакцина, снижая риск смерти на 22% (HR 0,78 [0,61–0,98]; p=0,032), продлевает жизнь на медианных 4,1 месяца относительно плацебо [10]. И это притом что за курс лечения из 3 доз необходимо выложить почти 200 тыс. долларов при отсутствии страхового медицинского покрытия [11].
С возрастом риск столкнуться с опоясывающим лишаем резко возрастает.
«Шингрикс» (Shingrix, RZV) авторства «ГлаксоСмитКляйн» (GlaxoSmithKline) — единственная вакцина, которая предоставляет безоговорочно мощную и надежную защиту от опоясывающего лишая.
После вакцинации «Шингрикс» иммунитет против опоясывающего лишая сохраняется на протяжении 10 лет и даже дольше.
Фармотрасль продолжает искать варианты улучшить защитные показатели «Шингрикс».
ЧТО ПРОИЗОШЛО
Иммунная защита от опоясывающего лишая, активированная вакциной «Шингрикс» (Shingrix, RZV), сохраняется на протяжении не менее чем десяти лет после прививки.
«Шингрикс» — вакцина, предназначенная для профилактики опоясывающего герпеса и постгерпетической невралгии.
Сила превентивной защиты, предоставляемой «Шингрикс», близка к 90% (или даже превышает) и держится многие годы, причем вне зависимости от возраста (справедливо для лиц без ослабленного иммунитета).
Инактивированная рекомбинантная вакцина «Шингрикс», разработанная «ГлаксоСмитКляйн» (GlaxoSmithKline), вводится внутримышечно дважды — с интервалом от двух до шести месяцев.
До появления «Шингрикс» профилактика опоясывающего лишая была возможна только одной вакциной: живой аттенуированной «Зоставакс» (Zostavax, ZVL) авторства «Мерк и Ко» (Merck & Co.). Иммунизационная эффективность «Зоставакс» посредственна: она снижается риск развития заболевания на 51%, а постгерпетической невралгии на 67%.
ПОЧЕМУ ЭТО ВАЖНО
У переболевших (обычно в детстве) ветряной оспой (ветрянкой) вызвавший ее вирус остается персистировать в нейронах спинномозговых узлов и черепных нервов. В случае реактивации латентного (спящего) вируса ветряной оспы (герпесвирус человека типа 3, Human alphaherpesvirus 3; варицелла-зостер, Varicella zoster virus, VZV) возникает другая форма этой инфекции — опоясывающий лишай (опоясывающий герпес).
Вероятность развития опоясывающего лишая возрастает по мере снижения Т-клеточного иммунитета, поэтому в группе риска находятся люди пожилого возраста или с ослабленной иммунной системой [1], а также те, кто страдает хроническими заболеваниями, такими как хроническая обструктивная болезнь легких (ХОБЛ), сахарный диабет или астма [2].
Заболевание, так или иначе поражающее каждого третьего человека [3] [4] [5] [6], приводит к болевому синдрому (жжение, покалывание, онемение, пульсация, повышенная чувствительность, пульсация) и появлению волдырей на коже с дерматомным типом распределения [7] [8] [9] [10].
Сыпь при опоясывающем лишае обычно проходит через две–четыре недели, тогда как обременяющая постгерпетическая невралгия у одной пятой заболевших может сохраняться месяцами и годами после заживления кожных поражений.
Среди множества других осложнений: офтальмологический опоясывающий герпес, синдром Рамсея Ханта (ганглионит узла коленца лицевого нерва), миелопатия, асептический менингит, энцефалит, полинейропатия (включая синдром Гийена — Барре), полирадикулит, васкулопатия, острый некроз сетчатки, прогрессирующий некроз наружных слоев сетчатки.
Лечение опоясывающего лишая, которое следует начинать не позднее 72 часов после манифестации заболевания, предполагает применение системных противовирусных препаратов, таких как валацикловир (valaciclovir), фамцикловир (famciclovir), ацикловир (aciclovir), бривудин (brivudine), фоскарнет (foscarnet), аменамевир (amenamevir).
КАК ЭТО РАБОТАЕТ
«Шингрикс» (Shingrix, GSK1437173A, Hz/su) — инактивированная рекомбинантная субъединичная вакцина, сочетающая антиген в виде оболочечного гликопротеина E (gE) вируса ветряной оспы и адъювантную систему AS01B, улучшающую иммунологический ответ [1] [2] [3].
Иммунное воздействие белка gE стимулирует выработку антител против него, тем самым формируя адаптивный иммунитет против вируса.
Фирменный адъювант AS01B, заключенный в липосомальную рецептуру, комбинирует 3-О-дезацилированный 4′-монофосфорил-липид A (MPL) сальмонеллы (Salmonella minnesota) и молекулу сапонина (QS-21) из растительного экстракта квиллайи мыльной (Quillaja saponaria).
MPL, работающий как агонист толл-подобного рецептора 4 (TLR), стимулирует антигенпрезентирующие клетки посредством активации врожденного иммунитета. QS-21 действует как стимулятор путей врожденного иммунитета в моноцитах. Липосомальная формуляция нивелирует присущую QS-21 гемолитическую активность, а также усиливает презентацию антигена по сравнению с эмульсионной рецептурой AS02 [4] [5] [6] [7] [8] [9].
ДОСТУПНОСТЬ
Коммерческий дебют вакцины «Шингрикс» (Shingrix, RZV) состоялся в Канаде в середине октября 2017 года [1].
В США «Шингрикс» получила одобрение Управления по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) дважды. Вначале, в конце октября 2017 года, для профилактики опоясывающего лишая среди лиц в возрасте 50 лет и старше [2]. А затем, в конце июля 2021-го, — для такой же задачи среди лиц в возрасте 18 лет и старше, находящихся в группе риска развития опоясывающего лишая ввиду иммунодефицита или иммуносупрессии по причине заболевания или терапии [3].
В Европе «Шингрикс» заручилась аналогичными маркетинговыми разрешениями Европейского агентства по лекарственным средствам (EMA) в конце марта 2018 года и конце марта 2020-го [4].
В России «Шингрикс» прошла регистрацию Минздрава по указанным двум показаниям в конце августа 2023 года [5].
БОЛЬШИЕ ДЕНЬГИ
Если в 2022 году «ГлаксоСмитКляйн» (GlaxoSmithKline) наторговала вакциной «Шингрикс» (Shingrix, RZV) на 2,96 млрд фунтов (3,67 млрд долларов), то в 2023-м ее реализация составила 3,45 млрд фунтов (4,27 млрд долларов).
[membership level=»2″ show_noaccess=»true»]
Перешагнуть планку в 1 млрд долларов продаж «Шингрикс» сумела мгновенно: через год после своего запуска.
Впечатляющий спрос на «Шингрикс» не идет ни в какое сравнение со скромными успехами вакцины «Зоставакс» (Zostavax, ZVL), которая, дебютировав в мае 2006 года, так и не смогла стать бестселлером: пик интереса пришелся на 2014 год, когда она принесла «Мерк и Ко» (Merck & Co.) 765 млн долларов.
Ввиду категорически превосходящей эффективности «Шингрикс» закат «Зоставакс» был неминуем. В середине ноября 2020 года «Мерк и Ко» прекратила ее продвижение в США, на главном рынке сбыта любой фармацевтической продукции [1].
История имеет и обратную сторону. В свое время «Мерк и Ко» и ее «Гардасил» / «Гардасил 9» (Gardasil / Gardasil 9) вытеснили «ГлаксоСмитКляйн» и ее «Церварикс» (Cervarix) из американского бизнеса прививок против вируса папилломы человека (ВПЧ). Так, если в 2023 году мировые продажи первых вышли к внушительным 3,94 млрд долларов, то второй — к скромным 117 млн фунтов (145 млн долларов).
[/membership]
ЭФФЕКТИВНОСТЬ
Клинические исследования ZOE-50 (ZOSTER-006, NCT01165177) и ZOE-70 (ZOSTER-022, NCT01165229) фазы III (рандомизированные, двойные слепые, плацебо-контролируемые, многоцентровые, международные), привлекшие свыше 31 тыс. человек соответственно в возрасте ≥ 50 и ≥ 70 лет, установили, после наблюдений в течение медианных 3,1 и 4,0 лет, следующую высокую профилактическую эффективность «Шингрикс» (Shingrix, RZV) против опоясывающего лишая [1] [2] [3] [4]:
≥ 50 лет: 97% (95% ДИ [здесь и далее]: 94–99);
≥ 60 лет: 98% (93–100);
≥ 70 лет: 91% (87–95);
≥ 80 лет: 91% (80–97).
«Шингрикс» аналогично успешно предотвратила развитие постгерпетической невралгии:
≥ 50 лет: 100% (77–100);
≥ 60 лет: 100% (41–100);
≥ 70 лет: 89% (69–97);
≥ 80 лет: 71% (−52–97).
«Шингрикс» защитила от развития осложнений опоясывающего лишая, отличных от постгерпетической невралгии, таких как васкулит, инсульт, а также диссеминированное, офтальмологическое, неврологическое или висцеральное заболевание. Профилактическая эффективность составила 94% (60–100) и 92% (43–100) среди лиц в возрасте ≥ 50 и ≥ 70 лет соответственно [5].
[membership level=»3″ show_noaccess=»true»]
«Шингрикс» надежно предотвращала развитие опоясывающего лишая на протяжении длительного времени после иммунизации, успешно преодолевая проблему старения иммунной системы и обеспечивая стойкую защиту вне зависимости от возраста. Так, после иммунизации «Шингрикс» превентивный щит поддерживался как минимум в течение 3 лет после прививки лиц в возрасте ≥ 50 лет [6] [7]. Эффективность вакцины на протяжении медианных 7 лет составила 91% (88–93), притом что уровень защиты изменялся следующим образом: 98% → 93% → 92% → 90% → 85% → 85% → 84% [8].
Для сравнения: эффективность вакцины «Зоставакс» (Zostavax, ZVL) для защиты от опоясывающего герпеса лиц в возрасте ≥ 50 лет ослабевала быстро, составляя 68% (65–70) в первый год после прививки, 47% (44–50) в течение второго года и 32% (15–45) по прошествии восьми лет [9].
Долгосрочные наблюдения за участниками (n>7000) клинического испытания ZOSTER-049 (NCT05371080) фазы III (рандомизированного, открытого, многоцентрового, международного) выяснили, что после вакцинации «Шингрикс» иммунитет против опоясывающего герпеса сохранялся очень долгое время на прилично высоком уровне [10]. Показатели устойчивой защитной эффективности таковы:
≥ 50 лет: 80% (74–85) — в период от 6 до 11 лет после прививки;
≥ 50 лет: 82% (63–92) — на 11-м году после вакцинации;
≥ 70 лет: 73% (63–81) — в период от 6 до 11 лет после прививки.
Клинические исследования ZOSTER-002 (NCT01610414; #1) и ZOSTER-039 (NCT01767467; #2) фазы III (рандомизированные, двойные слепые, плацебо-контролируемые, многоцентровые, международные) пригласили взрослых (18 лет и старше) пациентов (n=1721 и n=515) с ослабленной иммунной системой (иммунокомпрометированным статусом), — соответственно прошедших аутологичную трансплантацию гемопоэтических клеток или с гематологическими злокачественными новообразованиями (множественная миелома, неходжкинская B-клеточная лимфома, хронический лимфоцитарный лейкоз и др.) [11] [12].
Защитная эффективность «Шингрикс» составила:
≥ 18 лет (#1): 68% (55–78);
≥ 50 лет (#1): 67% (53–78);
≥ 18 лет (#2): 87% (44–99).
Эффективность «Шингрикс» в задаче предупреждения постгерпетической невралгии следующая:
≥ 18 лет (#1): 89% (23–100);
≥ 50 лет (#1): 88% (10–100).
Клиническое исследование NCT02581410 фазы III (нерандомизированное, открытое, многоцентровое) подтвердило, что «Шингрикс» успешно справляется с профилактикой опоясывающего лишая среди лиц, ранее привитых альтернативной вакциной «Зоставакс» [13] [14].
Клинические исследования NCT01954251, NCT02045836, NCT03439657 и NCT02052596 фазы III (рандомизированные, открытые, многоцентровые, многоцентровые, международные) продемонстрировали, что «Шингрикс» можно без каких-либо проблем назначать совместно с другими вакцинами, такими как противогриппозная «Флуарикс» (Fluarix), пневмококковые «Пневмовакс 23» (Pneumovax 23) и «Превенар 13» (Prevnar 13), а также «Бустрикс» (Boostrix) против столбняка, дифтерии и коклюша — не обнаружено признаков взаимного вмешательства в иммунный ответ со стороны антигенов, содержащихся в этих вакцинах [15] [16] [17] [18]. Параллельную вакцинацию уместно осуществлять инъекциями в разные анатомические места [19] [20].
Клиническое исследование ZOSTER-026 (NCT01751165) фазы III (рандомизированное, открытое, многоцентровое, международное) показало, что вторую (бустерную) дозу «Шингрикс» можно вводить равно как через 2 месяца после первой, так и по прошествии 6 месяцев [21].
[/membership]
АЛЬТЕРНАТИВЫ
С октября 2017 года в Южной Корее доступна вакцина «Скайзостер» (SKYZoster), разработанная местной «Эс-кей байосайенсиз» (SK Bioscience) для профилактики опоясывающего лишая у лиц в возрасте 50 лет и старше [1].
[membership level=»2,3″ show_noaccess=»true»]
Защитная эффективность живой аттенуированной вакцины «Скайзостер» (NBP608) находится на приблизительно таком же уровне, как у «Зоставакс» (Zostavax, ZVL) авторства «Мерк и Ко» (Merck & Co.) [2].
В 2022 году «Скайзостер» занимала 54% корейского рынка, оставшиеся 46% принадлежали «Зоставакс» [3].
Однако в 2023-м, в первый год доступности «Шингрикс» (Shingrix, RZV), всё резко поменялось: вакцина «ГлаксоСмитКляйн» (GlaxoSmithKline) мгновенно захватила 44% продаж, оставив «Скайзостер» и «Зоставакс» по 30% и 26%. Что примечательно, клиническому внедрению «Шингрикс» не помешала ее относительно высокая цена в 500 тыс. вон (365 долларов) — против стоимости «Скайзостер» в 150 тыс. вон (110 долларов) и «Зоставакс» в 170 тыс. вон (125 долларов) [4].
В январе 2023 года Китай одобрил первую вакцину для профилактики опоясывающего лишая собственной разработки. Живая аттенуированная вакцина создана «Чанчунь Би-си-эйч-ти байотекнолоджи» (Changchun BCHT Biotechnology) [5].
[/membership]
ТЕМ ВРЕМЕНЕМ
В начале 2024 года «Курево ваксин» (Curevo Vaccine), за которой стоит корейская «Си-джи байофарма» (CG Biopharma), уведомила об успешной клинической проверке амезосватеина (amezosvatein, CRV-101), экспериментальной адъювантной субъединичной вакцины на основе гликопротеина E (gE) вируса ветряной оспы, которая прошла прямое сравнение с «Шингрикс» (Shingrix, RZV) [1].
[membership level=»2,3″ show_noaccess=»true»]
В клиническом исследовании NCT05304351 фазы II вакцинный кандидат амезосватеин продемонстрировал равно как нехудшую профилактическую эффективность против опоясывающего лишая, так и улучшенный профиль безопасности.
Как утверждает «Курево», несмотря на высокий уровень защиты от опоясывающего герпеса, которым наделяет «Шингрикс», степень ее внедрения среди нуждающихся в этой прививке лиц остается низкой. Так, заявлено, что две трети американцев не иммунизированы против опоясывающего герпеса, тогда как в Европе, Китае и в целом в мире «Шингрикс» получили менее 5% представителей группы риска развития опоясывающего лишая. Проблема связана в том числе с переносимостью вакцины авторства «ГлаксоСмитКляйн» (GlaxoSmithKline): якобы после первой дозы «Шингрикс» приблизительно 20–30% человек не обращаются за второй ее дозой по причине нежелательных явлений (НЯ).
Согласно исследованиям самой «ГлаксоСмитКляйн», действительно после «Шингрикс» широко распространены такие НЯ, как боль (в среднем в 80% случаев), покраснение (38%) или отечность (27%) в месте инъекции, боль в мышцах (47%), усталость (47%), головная боль (40%), озноб (29%), повышение температуры тела (22%), тошнота, рвота, диарея или боль в области живота (18%) [2]. Однако нежелательные явления не приводили к столь внушительному, как уверяет «Курево», проценту отказа от второй дозы «Шингрикс» [3] [4].
[/membership]
КОНВЕЙЕР
Фармпроизводители не оставляют надежду предложить новые профилактические вакцины, которые бы эффективнее, чем «Шингрикс» (Shingrix, RZV), справлялись с предупреждением опоясывающего лишая и его осложнений.
[membership level=»2,3″ show_noaccess=»true»]
В четвертом квартале 2024 года китайская «Бэйцзин Лучжу байотекнолоджи» (Beijing Luzhu Biotechnology) намеревается отправить на регистрацию LZ901 — рекомбинантную вакцину, сделанную на основе гликопротеина E (gE) вируса ветряной оспы в тетрамерном исполнении и подкрепленную классическим адъювантным гидроксидом алюминия [1] [25].
Китайская «Цзянсу Рекбайо текнолоджи» (Jiangsu Recbio Technology) работает над REC610 — рекомбинантной gE-вакциной с фирменным адъювантом BFA01, который позиционируется усовершенствованной версией адъюванта AS01B в «Шингрикс» [2] [3] [4] [5].
Китайская «Кансино байолоджикс» (CanSino Biologics) совместно с британской «Баринтус байотерапьютикс» (Barinthus Biotherapeutics), ранее называвшей «Вакситек» (Vaccitech), трудится над VTP-400 (ChAdOx1-VZV, CS-2032, CSB-016) — вакциной на базе аденовирусного вектора шимпанзе (ChAdOx1), кодирующего антиген gE [6].
Китайская «Имморна байотекнолоджи» (Immorna Biotechnology) верит в успех JCXH-105 — вакцины на базе самореплицирующейся РНК (срРНК), кодирующей антиген gE [7] [8] [9].
Китайская «Максвакс байотекнолоджи» (Maxvax Biotechnology) исследует рекомбинантную gE-вакцину с фирменным адъювантом MA105, составленным из полиинозиновой:полицитидиловой кислоты (Poly I:C), функционирующей как агонист толл-подобного рецептора 3 (TLR3), молекулы сапонина (QS-21) из растительного экстракта квиллайи мыльной (Quillaja saponaria), липидных молекул [10] [11]
Корейская «Си-эйч-эй ваксин рисёрч инститьют» (CHA Vaccine Research Institute) изучает CVI-VZV-001 — рекомбинантную gE-вакцину с фирменным адъювантом L-pampo, который представляет собой агонист толл-подобных рецепторов 1/2 и 3 (TLR1/2 и TLR3) [12] [13] [14].
Корейская «Эубайолоджикс» (EuBiologics) сделала ставку на рекомбинантную вакцину, нанолипосомальные частицы которой демонстрируют иммунной системе антиген gE наряду с адъювантами EcML и CoPoP, представляющими собой соответственно монофосфорил-липид A (MPL) кишечной палочки (Escherichia coli) и кобальт-порфирин-фосфолипид (CoPoP) [15].
Корейская «ДженУан лайф сайенс» (GeneOne Life Science) прорабатывает ДНК-вакцину GLS-5100 (VGX-5100). Лежащая в ее основе ДНК-плазмида кодирует гликопротеин E (gE) и предранние белки IE63 и IE62 вируса ветряной оспы, а также адъюванты интерлейкин 7 (IL-7) и интерлейкин 33 (IL-33) [16] [17].
«Дайнавакс текнолоджис» (Dynavax Technologies) занимается клинической проверкой Z-1018 — рекомбинантной gE-вакцины, подкрепленной адъювантом CpG 1018, который является агонистом толл-подобного рецептора 9 (TLR9), и опциональным гидроксидом алюминия [19].
Не забыта, разумеется, мРНК-технология: «Байонтек» (BioNTech) и «Пфайзер» (Pfizer) тестируют BNT167, «Модерна» (Moderna) пробует силы с mRNA-1468, академические учреждения Китая разработали «Зосал» (Zosal) и mgE@Syn-LNP. Все эти вакцины против опоясывающего лишая кодируют антиген gE [20] [21] [22] [23] [24].
Ведущей экспериментальной онкологической вакциной является GT-30, изучаемая в иммуноонкологическом лечении распространенного рака печени (гепатоцеллюлярной карциномы).
Терапия второй линии, осуществленная после первоочередного назначения стандартных «Нексавара» (Nexavar, сорафениб) или «Ленвимы» (Lenvima, ленватиниб), вывела приличную пропорцию пациентов с неоперабельным или метастатическим раком печени к статусу ремиссии, тем самым открыв возможность для проведения потенциально излечивающего хирургического вмешательства наряду с облучением.
С позиции продления жизни пациентов клиническая результативность онковакцины GT-30 превзошла все одобренные фармакологические подходы к лечению распространенного рака печени.
В целом ситуация медленно, но верно улучшается. Так, если четыре десятка лет назад пятилетняя относительная выживаемость при раке печени в США составляла крошечных 3%, сейчас этот показатель подрос до 22% [1]. Но этого решительно недостаточно: данный показатель идет вслед за относительной выживаемостью в ничтожных 13% при раке поджелудочной железы [2].
Как бы то ни было, несмотря на впечатляющий прогресс медицинской науки, говорить о победе над раком печени еще очень и очень рано.
В реальности нет смысла лечить неоперабельный рак печени препаратами.
ПОЧЕМУ ЭТО ВАЖНО
Гепатоцеллюлярная карцинома, будучи наиболее распространенной формой первичного рака печени [1], является одной из основных причин смерти от онкологических заболеваний во всём мире [2]. Ежегодно ставится свыше 860 тыс. диагнозов рака печени и умирает более чем 760 тыс. человек [3] [4]. В большинстве случаев гепатоцеллюлярная карцинома возникает на фоне цирроза печени и связана с определенными факторами риска, такими как вирусный гепатит B или C, чрезмерное употребление алкоголя, а также неалкогольная жировая болезнь печени (НАЖБП), ассоциированная с метаболическим синдромом и сахарным диабетом [5] [6] [7] [8]. Рак печени в 60% случаев выявляется не на ранней стадии, не подходящей для проведения оперативного вмешательства, которое в теории способно его вылечить [9] [10] [11] [12]. Существующие лекарственные препараты против распространенной гепатоцеллюлярной карциномы характеризуются весьма скромной терапевтической эффективностью, по сути не предоставляющей пациентам какого-либо ощутимого шанса остаться в живых [13] [14] [15] [16].
ПОДХОД
«Джинеос терапьютикс» (Geneos Therapeutics), будучи дочерним биотехнологическим стартапом «Иноувио фармасьютикалс» (Inovio Pharmaceuticals), официально появилась в поле зрения инвесторов в феврале 2019 года. На сегодня «Джинеос» привлекла стороннее финансирование в размере 44,5 млн долларов [1] [2].
Если материнская «Иноувио» погружена в «популярные» заболевания вроде дисплазий, вызванных вирусом папилломы человека (ВПЧ), или коронавирусной инфекции COVID-19, то «Джинеос» сделала ставку на те патологии, к которым весьма трудно подобраться ввиду явных сложностей с лечением. Сейчас стартап сосредоточен на лечении двух онкологических состояний: распространенной гепатоцеллюлярной карциномы и впервые диагностированной глиобластомы с неметилированным промотором MGMT.
«Джинеос» лицензировала у «Иноувио» фирменные технологии создания ДНК-вакцин и их доставки в организм.
В рамках процесса SynCon генерируется ДНК-последовательность, несущая отобранные антигены и оптимизированная в целях улучшения стабильности мРНК, загрузки рибосом и экспрессии антигенных белков. Вакцинный препарат, представляющий собой плазмиды, в которые встроена готовая ДНК-последовательность, внутрикожно или внутримышечно вводится в организм.
Для того чтобы плазмиды проникли в клетки используется устройство «Селлектра» (Cellectra), организующее электропорацию, когда ряд кратковременных электрических импульсов заставляет клеточные мембраны открываться. Через несколько часов или дней клетки начинают синтезировать антигены — их презентирование иммунной системе запускает продуцирование антител и T-киллерных клеток, «заряжая» организм возможностью борьбы с опухолевыми клетками.
«Джинеос» объединила эти технологии в лице персонализированной иммунотерапевтической платформы GT-EPIC, на рельсах которой организовано создание специфичной для каждого пациента вакцинной ДНК-плазмиды, кодирующей неоантигены (ненормальные мутации или вариации генома, продуцируемые раковыми клетками), полученные из РНК опухолевых биоптатов.
Онковакцина затем сочетается с другой ДНК-плазмидой, кодирующей цитокин интерлейкин 12 (IL-12), в данном случае выступающий адъювантом. Комбинированная молекула внутрикожно доставляется в организм электропорационным устройством «Селлектра». Иммуноонкологический терапевтический коктейль активирует неоантигеноспецифические T-хелперы CD4+ и киллерные T-клетки CD8+, целенаправленно уничтожающие опухолевые клетки.
Персонализированная иммунотерапия обеспечила уверенное продление жизни для трети пациентов с протоковой аденокарциномой поджелудочной железы.
СИЛЬНЫЕ СТОРОНЫ
«Джинеос» уверена, что платформа GT-EPIC располагает рядом преимуществ перед решениями конкурентов, занимающихся персонализированным противораковым лечением, таким как неоантигенные вакцины и клеточная терапия.
Во-первых, плазмиды стимулируют ответы со стороны T-клеток CD4+ и CD8+, что характерно для вакцин на основе вирусных векторов, но уже сложнее для вакцин на базе белков или пептидов. Связано это с тем, что и плазмиды, и вирусные векторы вызывают экспрессию антигена внутри клеток: для генерации устойчивого и сильного T-клеточного CD8+ ответа антигенам требуется быть презентированными на молекулах класса I главного комплекса гистосовместимости (MHC-I), то есть процессинг и презентирование должно происходить внутриклеточно.
Во-вторых, плазмиды способны нести относительно высокую полезную нагрузку: их емкость позволяет кодировать существенно большее число различных антигенов в сравнении с вирусными векторами. В продолжающихся клинических испытаниях «Джинеос» использует до 40 неоантигенов для каждого пациента, но, согласно доклиническим исследованиям, без особых проблем можно закодировать гораздо больше таковых (даже 80). Концепция состоит в том, чтобы демонстрировать иммунной системе пациента как можно большее число неоантигенов-мишеней — она сама разберется, какие из них будут лучше стимулировать иммунный ответ, по итогам отразившийся должной клинической эффективностью.
В отрасли ведутся дебаты, могут ли неоантигены с неоптимальным противоопухолевым эффектом блокировать более продуктивные неоантигенные ответы посредством иммунодоминирования. «Джинеос», опирающаяся на клинические данные, придерживается позиции, что пациенты, получившее большее количество неоантигенов, отвечают на лечение лучше, чем получившее меньшее их число.
Если сравнивать ДНК-вакцины с мРНК-вакцинами, последние всё же оптимальнее с позиций доставки полезной нагрузки: им необходимо лишь пересечь плазматическую мембрану, дабы запустить производство белковых антигенов, тогда как первые должны дополнительно миновать ядерную оболочку, что снижает итоговую эффективность. Тем не менее преимущество ДНК-вакцин состоит в их большей стабильности (они могут годами храниться при температуре 2–8 ℃), то есть они практичнее с точки зрения дистрибуции в развивающихся странах и сельской местности, не располагающих должной холодовой инфраструктурой хранения готовых препаратов.
В-третьих, важным преимуществом платформы GT-EPIC является относительно низкая себестоимость продукции и более короткие сроки изготовления плазмид, если сравнивать с вакцинами на основе аденовирусных векторов или мРНК либо CAR-T-клеточной терапией. Ценовой аспект позволит по-настоящему демократизировать персонализированное лечение рака.
Опять же, высокая скорость создания готовых плазмид, укладывающаяся в 6–8 недель (впоследствии процесс ужмется до 3–4 недель), важна для пациентов с раком на поздних стадиях: такие больные, ввиду стремительного прогрессирования онкологического заболевания, не в силах ждать 4–6 месяцев, когда будет готов индивидуализированный лекарственный препарат. Оперативность подготовки плазмид также открывает возможность для лечения онкопатологии на ранних стадиях, не прибегая к хирургическому вмешательству.
НАЧАЛО
GT-30, ведущая экспериментальная противораковая вакцинная программа «Джинеос», ориентирована на лечение распространенной гепатоцеллюлярной карциномы посредством вышеописанной иммунотерапии, сочетанной с «Китрудой» (Keytruda, пембролизумаб), блокатором PD-1 авторства «Мерк и Ко» (Merck & Co.).
Рак печени был выбран по той причине, что в его «холодных» опухолях отсутствуют инфильтрирующие лимфоциты, то есть опухолевые клетки решительно недостаточно отвечают на назначение только ингибиторов PD-(L)1.
Применение GT-30 превращает опухоли в «горячие» иммуногенные путем примирования (запуска) ответа T-клеток на периферии, вдали от опухоли. Терапия преодолевает определенные ключевые механизмы резистентности, задействованные при раке печени, фактически «загоняя» T-клетки в опухолевые клетки.
В ноябре 2022 года «Джинеос» продемонстрировала, что применение GT-30 обеспечило частоту контроля над заболеванием (DCR) у 54% (n=13/24) пациентов, причем 13% (n=3/24) человек достигли полного ответа (CR), то есть вышли к ремиссии [1].
Для сравнения: CR при лечении рака печени только «Китрудой» не превышает 4%.
Согласно анализу репертуара T-клеточных рецепторов (TCR) в периферической крови и опухолевой ткани, осуществленному до и после иммуноонкологической вакцинации, у всех пациентов выявлены новые или размножившиеся клоны T-клеток, преимуществено с активированным фенотипом CD8+. К 9-й неделе лечения они проникали в микроокружение опухоли (TME), что, скорее всего, способствовало ее регрессии.
СЕЙЧАС
Клиническое исследование GT-30 (NCT04251117) фазы Ib/IIa (нерандомизированное, открытое, многоцентровое) охватило взрослых пациентов (n=36) с гепатоцеллюлярной карциномой, которые ранее получили первоочередное лечение тирозинкиназными ингибиторами «Нексаваром» (Nexavar, сорафениб) или «Ленвимой» (Lenvima, ленватиниб), в ответ на которое либо заболевание прогрессировало, либо развилась непереносимость.
Неоперабельная гепатоцеллюлярная карцинома испытуемых должна была находиться, согласно Барселонской системе стадирования рака печени (BCLC), либо на стадии C (распространенная стадия), либо на стадии B (промежуточная стадия), не подходящей для проведения локорегионарной терапии или рефрактерной к ней.
На момент анализа данных, который состоялся по прошествии наблюдений в течение медианных 21,5 месяца, 34 из 36 пациентов прошли хотя бы одно повторное сканирование, то есть для них имелась возможность оценить ответ на лечение в соответствии с критериями RECIST 1.1.
Два пациента прекратили терапию по причине несвязанных с ней тяжелых нежелательных явлений (НЯ), но были включены в полный анализ данных в качестве нереспондентов.
Частота общего ответа (ORR) составила 31% (n=11/36). К полному ответу (CR), то есть к статусу ремиссии рака печени, вышли 8% (n=3/36) испытуемых [1] [2] [3].
Дополнительно четвертому больному, заболевание которого изначально было неоперабельным, удалось полностью избавиться от всех опухолевых поражений (первичных в печени и метастазных в легких) — после пятой дозы терапевтической персонализированной противораковой вакцины, что открыло для него возможность пройти потенциально излечивающую хирургическую резекцию вкупе с облучением.
Частичный ответ (PR) был получен у 22% (n=8/36) человек, стабилизация заболевания (SD) была зафиксирована у 25% (n=9/36) испытуемых, с прогрессированием рака печени столкнулись 39% (n=14/36) участников.
Таким образом, частота контроля заболевания (DCR), как сумма CR, PR и SD, составила 56% (n=20/36).
Подтверждена оправданность использования ультрачувствительного анализа третьего поколения, оценивающего уровень циркулирующей опухолевой ДНК (ctDNA) и позволяющего относительно надежно установить текущий статус заболевания (прогрессирование или регрессирование).
Анализ ctDNA характеризуется высокой прогностической значимостью: снижение уровня ctDNA при раке печени обычно предшествует улучшению результатов на МРТ, тем более что очаги гепатоцеллюлярной карциномы, если судить по МРТ, зачастую не разрешаются полностью, несмотря на отсутствие клинических признаков остаточного заболевания.
Медиана длительности ответа (DOR) не достигнута, медиана выживаемости без прогрессирования (PFS) составила 4,2 месяца, медиана общей выживаемости (OS) получилась равной 19,9 месяца.
Экспериментальное лечение характеризовалось приемлемым профилем безопасности, в целом не отличающимся от применения только пембролизумаба. Серьезных НЯ после назначения противораковой вакцины зарегистрировано не было. Наиболее частыми НЯ были реакции по месту инъекций, которые носили преходящий характер легкой степени тяжести.
мРНК-технологии рисуют оптимистичное будущее в лечении онкологических заболеваний.
СУТЬ
Наблюдаемый в настоящее время научный прогресс в области разработки и клинической противораковых мРНК-вакцин характеризуется тем, что такие препараты применяются почти исключительно в адъювантном режиме: для предотвращения рецидива после хирургического удаления опухоли. И потому складывается мнение, будто терапевтические персонализированные онковакцины эффективны лишь после резекции, но никак не до ее проведения, то есть они якобы не работают при лечении распространенных, неоперабельных или метастатических опухолей. Но это ошибочно, если судить по обнадеживающим результатам применения ДНК-вакцины GT-30 авторства «Джинеос», которая наделила полными ответами пациентов с поздними стадиями прогрессирующего рака печени, который хирургическое вмешательство не проходил.
ИТОГИ
Лечение распространенной гепатоцеллюлярной карциномы при помощи GT-30, терапевтической персонализированной противораковой вакцины, нашло ответ у трети пациентов (ORR 31%), и это весьма приличный показатель.
Согласно ряду клинических испытаний моноприменения блокаторов PD-(L)1, таких как «Китруда» (Keytruda, пембролизумаб), «Опдиво» (Opdivo, ниволумаб), «Имфинзи» (Imfinzi, дурвалумаб) и «Тевимбра» (Tevimbra, тислелизумаб), изученных в первоочередном и второлинейном лечении распространенного рака печени, выдаваемая ими ORR укладывалась в пределы 12–18% [1] [2] [3] [4] [5] [6] [7] [8].
Более того, GT-30 обеспечила полный ответ в 8% случаев и медиану общей выживаемости на уровне 19,9 месяца, тогда как указанные ингибиторы иммунных контрольных точек (ИИКТ) вывели эти показатели к более скромным диапазонам в 2–4% и 13,2–16,6 месяца. Однако онковакцина не смогла существенным образом сдержать прогрессирование заболевания.
Моноприменение блокаторов PD-(L)1 практически не в силах выдать какой-либо полный ответ при гепатоцеллюлярной карциноме ввиду иммунной резистентности, обусловленной спецификой опухолевого микроокружения, и низкой опухолевой мутационной нагрузки (TMB) [9].
Зато персонализированная иммунотерапия, благодаря своей способности индуцировать опухолеспецифические T-клеточные ответы, сенсибилизирует опухоли к назначению ИИКТ, которые, «снимая тормоза» с иммунной системы, начинают работать должным образом [10] [11] [12] [13] [14] [15].
В целом состоятельность подхода, организованного «Джинеос», неоднократно подтверждалась разработчиками других терапевтических персонализированных противораковых вакцин: как в доклинических исследованиях [16] [17] [18] [19], так и в клинических испытаниях при меланоме, глиобластоме, раке поджелудочной железы, немелкоклеточном раке легкого, рак мочевого пузыря [20] [21] [22] [23] [24] [25] [26]. Однако именно «Джинеос» одной из первых уверенно доказала, что метод эффективно работает в случае гораздо менее чувствительных к иммунотерапии типах опухолей, таких как гепатоцеллюлярная карцинома.
Монотерапия моноклональными антителами против PD-(L)1, как известно, обращает вспять дисфункцию T-клеток в существующих неоантиген-специфических T-клеточных клонах, но не может индуцировать новые неоантиген-специфические T-клеточные клоны [27] [28].
Согласно иммунологическому анализу, добавление онковакцины GT-30 устранило это досадное ограничение, обеспечив как индукцию новых Т-клеточных ответов на кодируемые ею неоантигены, так и расширение репертуара T-клеточных рецепторов (TCR) в периферической крови и опухоли. Согласно анализу секвенирования отдельных клеток, основной вклад в рост T-клеточного пула был сделан со стороны эффекторных T-клеток памяти CD8+.
«Джинеос» не устает подчеркивать оправданность включения в терапевтическую онковакцину как можно большего числа различных целевых неоантигенов, не обходя стороной драйверные (driver) и пассажирские (passenger), основные (truncal) и дополнительные (branch), общие (shared) и частные (private) мутации, а также неоантигенные эпитопы с более широким диапазоном прогнозируемой аффинности связывания с молекулами I класса человеческого лейкоцитарного антигена (HLA). Максимально развернутый репертуар неоантигенов, доставляемых онковакциной, приводит к задействованию более широкого набора иммунных реакций, тем самым улучшая шансы на успешное лечение.
Попутно высказана гипотеза, что сопутствующие факторы, такие как микроокружение опухоли и иммунная приспособленность, могут играть определяющую роль в характере клинического ответа [29].
Ограничением онковакцины GT-30 является возможное раннее приобретение резистентности к ней вследствие неоантигенной потери и гетерогенности опухоли. Решением может стать сбор образцов опухолевой ДНК из нескольких очагов в сочетании с быстрым синтезом новых версий персонализированной вакцины в целях возвращения контроля над очагами, не реагирующими на терапию.
ПЕРСПЕКТИВЫ
Методы лечения неоперабельного рака печени стремительно развиваются, и потому, очевидно, в обозримом будущем появятся такие, которые обеспечат улучшенные клинические исходы, чем онковакцина GT-30.
Так, например, уже одобрены иммунотерапевтические комбинации из атезолизумаба (atezolizumab) и бевацизумаба (bevacizumab), дурвалумаба (durvalumab) и тремелимумаба (tremelimumab), камрелизумаба (camrelizumab) и ривоцераниба (rivoceranib), на которые пациенты весьма неплохо отвечают [1] [2] [3].
Однако у персонализированного лечения гепатоцеллюлярной карциномы, предложенного «Джинеос», есть неоспоримое преимущество, связанное с высокой безопасностью. Конкурирующие терапевтические подходы, напротив, характеризуются повышенным риском развития тяжелых или серьезных иммуноопосредованных нежелательных явлений, то есть для их применения подходят далеко не все пациенты. Вот почему онковакцина GT-30 непременно займет должное место в арсенале лечения рака печени.
БУДУЩЕЕ
Как и любое новое направление медицины, терапевтические персонализированные противораковые вакцины проходят длинный путь разработки и развития, сопровождающийся своими падениями и взлетами.
Эволюция в этой области напоминает историю нынешней иммунотерапии онкологических заболеваний, которая, без оглядки на обилие неудач в ходе экспериментов, в итоге всё же подарила надежду пациентам с солидными и гематонкологическими опухолями. Так, на протяжении почти 20 лет многочисленные клинические испытания моноклональных антител не демонстрировали воспроизводимой эффективности, прежде чем в 1997 году на сцену не вышел ритуксимаб (rituximab) [1]. Долгие годы блокаторы PD-(L)1 не показывали ощутимой клинической результативности — пока в 2008 году не были опубликованы первые результаты применения ниволумаба (nivolumab) [2]. В течение многих лет у CAR-T-клеток не получалось встать на рельсы успешной терапевтической модальности [3].
Онковакцины, индивидуализированные с учетом опухолевой специфики конкретного пациента, засвидетельствовали равно как собственное механистическое обоснование, так и убедительные клинические результаты. Они вплотную подошли к тому этапу становления, когда можно смело утверждать, что вскоре они примкнут к методам стандартной противораковой терапии.
Особенно преуспели «Модерна» (Moderna) и «Байонтек» (BioNTech), мРНК-онковакцины которых успешно прошли клиническую проверку лечения меланомы и рака поджелудочной железы соответственно.
Важнейшей темой для обсуждения остается выбор сроков проведения иммунотерапевтической вакцинации. Большинство персонализированных онковакцин исследуются на поздних стадиях заболевания и после применения ингибиторов иммунных контрольных точек (ИИКТ), однако, есть мнение, что они могут принести гораздо больше пользы, если будут подключаться на более ранних этапах терапии: например, в неоадъювантном периоде (до хирургического вмешательств), когда пациент еще не столкнулся с феноменом иммуноредактирования, истощением Т-клеток, дальнейшим ослаблением иммунной системы [4].
«Винревэйр» (Winrevair, сотатерцепт) — новый лекарственный препарат, предназначенный для лечения легочной артериальной гипертензии (ЛАГ) и способствующий повышению физической работоспособности, улучшению функционального класса заболевания и снижению риска клинических ухудшений.
Сотатерцепт (sotatercept), предложенный «Мерк и Ко» (Merck & Co.), нацелен на непосредственные патофизиологические механизмы ЛАГ, а не на купирование ее симптомов, чем занимаются все существующие на рынке лекарственные препараты, которые работают, по сути, как вазодилататоры, снимая гипертензию и, соответственно, нагрузку на сердце.
Сотатерцепт, представляющий собой слитый белок рецептора активина, действует совершенно иначе. Сотатерцепт, благодаря связыванию с активином А, уменьшает пролиферацию клеток в стенках легочной артерии и устраняет ригидность (жесткость) артерий, являющуюся одной из основных причин развития ЛАГ. В итоге сотатерцепту по силам изменить течение легочной артериальной гипертензии, а не просто временно устранить ее симптомы.
Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) одобрило «Винревэйр» в конце марта 2024 года [1].
Европейское агентство по лекарственным средствам (EMA) изучает заявку на регистрацию cотатерцепта.
Стоимость одного флакона «Винревэйр» для американских пациентов с ЛАГ составляет 14000 долларов. Поскольку препарат назначается подкожными инъекциями каждые три недели, годовой курс лечения обойдется в 238 тыс. долларов.
Согласно подсчетам Института клинико-экономической экспертизы США (ICER), экономический приемлемая стоимость годового курса лечения cотатерцептом американских пациентов с легочной артериальной гипертензии должна укладываться в диапазон от 17,9 до 35,4 тыс. долларов [2].
Пероральный препарат авторства Merck & Co. в пух и прах разбивает всех конкурентов для лечения и профилактики атеросклероза.
«Винревэйр»: механизм действия сотатерцепта
Легочная артериальная гипертензия (ЛАГ) — редкий тип легочной гипертензии, характеризующийся гемодинамическим и патофизиологическим состоянием повышенного среднего давления в легочной артерии (> 20 мм рт. ст. в покое) при отсутствии сопутствующей патологии легких или левосторонней сердечной недостаточности как причины повышения давления.
ЛАГ свойственны пролиферативное ремоделирование (изменение структуры) мелких легочных артерий и прогрессирующее сужение их просвета [1]. Возникающее при этом повышение давления в легочной артерии создает нагрузку на сердце, приводящую в конечном итоге к правожелудочковой недостаточности и смерти.
Существуют следующие фармакологические методы лечения легочной артериальной гипертензии [2] [3]:
ингибиторы фосфодиэстеразы типа 5 (PDE5i): силденафил (sildenafil), тадалафил (tadalafil);
При самостоятельном или комбинированном применении эти лекарственные средства улучшают легочную гемодинамику, повышают физическую работоспособность, замедляют прогрессирование ЛАГ. Однако данные препараты действуют главным образом как сосудорасширяющие средства, не оказывающие какого-либо значимого благотворного влияния на биологическую причину заболевания.
Медиана выживаемости при легочной артериальной гипертензии составляет от 5 до 7 лет после постановки диагноза, и за последнее десятилетие существенного улучшения этого показателя не произошло [4].
По-прежнему сохраняющиеся высокая заболеваемость и смертность подчеркивают необходимость разработки дополнительных методов лечения ЛАГ, направленных на новые сигнальные пути, участвующие в ремоделировании легочных сосудов.
Ремоделирование легочных сосудов, которое затрагивает все слои стенки сосуда, преимущественно обусловлено неконтролируемым усилением пролиферации и уменьшением апоптоза эндотелиальных и гладкомышечных клеток [5].
Согласно ряду исследований, существенный вклад в эти патофизиологические процессы вносит несбалансированная сигнализация со стороны членов суперсемейства трансформирующего фактора роста бета (TGF-β), как то: рецептор II типа костного морфогенетического белка (BMPR2), рецептор типа IIA активина A (ACVR2A, ActRIIA) и его лиганды, такие как активин А, активин В, фактор дифференцировки роста 8 (GDF8, миостатин) и фактор дифференцировки роста 11 (GDF11, BMP11). Сдвиг в сторону пролиферативной и антиапоптотической сигнализации, обеспеченный этими представителями суперсемейства TGF-β, считается одним из основных механизмов ремоделирования легочных сосудов у пациентов с легочной артериальной гипертензией [6] [7] [8].
Сотатерцепт (sotatercept, MK-7962, ACE-011) представляет собой первый в своем классе слитый белок, состоящий из Fc-домена человеческого иммуноглобулина G1 (IgG1), прилинкованного к модифицированному внеклеточному домену человеческого ACVR2A. Сотатерцепт действует как лигандная ловушка для отдельных представителей суперсемейства TGF-β.
Ингибирование определенных лигандов сотатерцептом приводит к восстановлению баланса гомеостаза легочных сосудов путем сдвига в сторону подавления как пролиферации, так и проапоптотической сигнализации. На животных моделях легочной гипертензии сотатерцепт ингибировал клеточную пролиферацию, способствовал апоптозу и снимал воспаление в стенках сосудов, что приводило к обращению вспять ремоделирования и восстановлению проходимости сосудов [8] [9] [10].
«Мерк и Ко» (Merck & Co.) получила сотатерцепт в ходе приобретения «Акселерон фарма» (Acceleron Pharma) за 11,5 млрд долларов, сделка по которому была закрыта в конце ноября 2021 года.
«Мерк и Ко» придется делиться доходами от реализации сотатерцепта (роялти 22%) с «Бристол-Майерс Сквибб» (Bristol-Myers Squibb), так как последняя купила «Селджен» (Celgene), которая ранее договорилась с «Акселерон» о совместной разработке этой молекулы и которая владела 11,5% этой фармкомпании [11] [12].
«Винревэйр»: эффективность и безопасность сотатерцепта
Опорное клиническое исследование STELLAR (NCT04576988) фазы III (рандомизированное, двойное слепое, плацебо-контролируемое, многоцентровое, международное) охватило взрослых пациентов (n=323) с легочной артериальной гипертензией (ЛАГ).
Основным критерием включения в испытание была документально подтвержденная диагностическая катетеризация правого сердца в любое время до скрининга, подтверждающая диагноз ЛАГ 1-й группы и симптоматической ЛАГ функционального класса II или III , согласно классификации Всемирной организации здравоохранения (ВОЗ), при любом из следующих подтипов ЛАГ: идиопатическая, наследственная, вызванная лекарственными препаратами или токсинами, связанная с заболеванием соединительной ткани, связанная с ликвидированными неосложненными врожденными системно-легочными шунтами, — исключая подтипы, связанные с порто-легочной патологией, шистосомозом, инфекцией вирусом иммунодефицита человека (ВИЧ) или вено-окклюзионной болезнью.
Участникам назначали подкожными инъекциями плацебо или сотатерцепт в целевой дозе 0,7 мг/кг каждые 3 недели — на фоне стандартной терапии ЛАГ.
По прошествии 24 недель лечения легочной артериальной гипертензии результаты получились следующими [1].
Медиана изменения в тесте на прохождение дистанции в течение 6 минут (6MWT) составила +34,4 м (95% ДИ [здесь и далее]: 33,0–35,5) в группе сотатерцепта — против +1,0 м (−0,3 — 3,5) в группе плацебо.
Согласно оценке Ходжеса — Лемана, разница результатов теста 6MWT, оценивающего физическую работоспособность, составила +40,8 м (27,5–54,1; p<0,001) в пользу сотатерцепта.
Пропорция пациентов, ответивших по всем трем критериям многокомпонентного улучшения: 38,9% испытуемых против 10,1% (p<0,001).
Речь идет об одновременном сочетании следующих показателей: улучшение результатов теста 6MWT хотя бы на 30 м; улучшение уровня N-концевого фрагмента мозгового натрийуретического пептида (NT-proBNP) — либо снижение минимум на 30%, либо поддержание существующего уровня, либо снижение ниже 300 пг/мл; улучшение функционального класса по классификации ВОЗ или его поддержание на уровне II класса. Выход к статусу многокомпонентного улучшения ассоциирован с более чем 50-процентным снижением относительного риска смерти [2].
Легочное сосудистое сопротивление (ЛСС) снизилось на 165,1 дин/с/см−5 (−176,0 — −152,0) — против увеличения на 32,8 дин/с/см−5 (26,5–40,0).
Согласно оценке Ходжеса — Лемана, разница в изменении ЛСС, отражающего тяжесть поражения артериального русла малого круга кровообращения, составила −234,6 дин/с/см−5 (−288,4 — −180,8; p<0,001) в пользу сотатерцепта.
Уровень NT-proBNP снизился на медианных 230,3 пг/мл (−236,0 — −223,0) — против роста на 58,6 пг/мл (46,0–67,0).
Согласно оценке Ходжеса — Лемана, разница в изменении уровня NT-proBNP, составила −441,6 пг/мл (−573,5 — −309,6; p<0,001) в пользу сотатерцепта.
Пропорция испытуемых, оказавшихся в статусе низкого риска ЛАГ, согласно критериям французского регистра пациентов с ЛАГ: 39,5% (31,9–47,5) — против 18,2% (12,6–25,1).
Под низким риском понимается соответствие всем трем следующим критериям: функциональный класс I или II по классификации ВОЗ, прохождение более чем 440 м в тесте 6MWT, уровень NT-proBNP ниже 300 пг /мл [3].
Пропорция пациентов, продемонстрировавших улучшение функционального класса по классификации ВОЗ: 29,4% (22,6–37,1) — против 13,8% (8,9–20,2).
Применение сотатерцепта обеспечило продление времени до смертельного исхода или нелетального ухудшения клинического состояния, снизив риск указанных событий на 82%: отношение риска (hazard ratio, HR) 0,16 (0,08–0,35).
Ухудшение клинического состояние подразумевает столкновение с любым из следующих событий: включение в список трансплантации легких и/или сердца; необходимость в резервном лечении ЛАГ-препаратами или необходимость в увеличении дозы инфузионного простациклина на 10% и более; необходимость в проведении предсердной септостомии; госпитализация; ухудшение функционального класса по классификации ВОЗ с одновременным ухудшением результатов прохождения теста 6MWD на 15% и более.
Назначение сотатерцепта привело к улучшению симптомов и последствий легочной артериальной гипертензии со стороны физических возможностей и сердечно-легочных функций, согласно пациентскому опроснику PAH-SYMPACT качества жизни при ЛАГ.
Применение сотатерцепта характеризовалось приемлемой переносимостью: только 1,8% испытуемых прервали лечение — против 6,2% в группе плацебо. Среди наиболее распространенных нежелательных явлений (НЯ), связанных с назначением сотатерцепта: эпистаксис (носовое кровотечение; у 20% пациентов), головокружение (15%), телеангиэктазия (стойкое расширение мелких сосудов кожи; 14%). Серьезные НЯ были редкими: только у 1,2% участников в обеих группах.
Что дальше
«Мерк и Ко» (Merck & Co.) продолжает клиническую проверку сотатерцепта (sotatercept), надеясь существенно расширить популяцию пригодных для его назначения пациентов.
Так, клиническое исследование ZENITH (NCT04896008) фазы III изучает лечение сотатерцептом более тяжелых пациентов с легочной артериальной гипертензией (ЛАГ). Если регистрационное STELLAR (NCT04576988) пригласило больных ЛАГ функционального класса II или III, согласно классификации Всемирной организации здравоохранения (ВОЗ), то это испытание охватывает участников с функциональным классом III и IV и высоким риском смерти. Выясняется, насколько продолжительно сотатерцепт отсрочивает наступление критического события, такого как смертельный исход, необходимость в легочной трансплантации или госпитализация.
Клиническое исследование HYPERION (NCT04811092) фазы III тестирует сотатерцепт среди пациентов с функциональным классом II или III и промежуточным или высоким риском прогрессирования ЛАГ: уточняется, продлевает ли он время до ухудшения клинического состояния.
Клиническое исследование MOONBEAM (NCT05587712) фазы II обкатывает сотатерцепт в педиатрической (1–17 лет) популяции пациентов с ЛАГ.
Продолжается клиническое исследование SOTERIA (NCT04796337) фазы III, необходимое для выяснения долгосрочных (6 лет) безопасности и эффективности сотатерцепта, назначаемого на постоянной основе.
Наконец, «Мерк и Ко» полагает, что сотатерцепт найдет применение в лечении комбинированной пре- и посткапиллярной легочной гипертензии, вызванной сердечной недостаточностью с сохраненной фракцией выброса. Для этого осуществляется клиническое исследование CADENCE (NCT04945460) фазы II.
Большие деньги
Нет никаких сомнений, что «Винревэйр» (Winrevair, сотатерцепт) ждут радужные бизнес-перспективы. В 2023 году мировой рынок лечения легочной артериальной гипертензии (ЛАГ) оценивался в 7,3 млрд долларов, а к 2032 году, согласно отраслевым прогнозам, он вырастет до 12,2 млрд долларов [1].
Сейчас половину рынка ЛАГ контролирует «Джонсон энд Джонсон» (Johnson & Johnson) своими препаратами «Аптрави» (Uptravi, селексипаг) и «Опсамит» (Opsumit, мацитентан), в 2023 году заработавшими 1,58 млрд долларов и 1,97 млрд долларов, что на 11% и 20% больше, чем в 2022-м.
С учетом того ассортимента масштабных благотворных терапевтических эффектов, которые оказывает сотатерцепт на течение ЛАГ и почти что не оказывают все прочие лекарственные средства, фокус врачей и пациентов будет всё больше и чаще смещаться именно в его сторону.
Согласно оптимистичным отраслевым прогнозам, максимальный годовой объем продаж, к которому может выйти «Винревэйр», составляет 7,5 млрд долларов. Более сдержанные предсказания указывают на суммы в промежутке от 3 до 4 млрд долларов. Сама «Мерк и Ко» (Merck & Co.) говорит обтекаемо, уверяя, что сотатерцепт станет «многомиллиардным лекарством».
Оптимистичное будущее
Кардиометаболический бизнес — новое для «Мерк и Ко» (Merck & Co.) направление деятельности, которое, судя по оптимистичным настроениям американского фармацевтического гиганта, сможет выйти к 15 млрд долларов продаж к середине 2030-х годов благодаря усилиям по разработке новых лекарственных средств.
Сейчас кардиометаболическая деятельность «Мерк и Ко» представлена единственным коммерциализированным препаратом — «Веркуво» (Verquvo, верицигуат). Этот пероральный стимулятор растворимой гуанилатциклазы (sGC), разработанный совместно с «Байер» (Bayer) и дебютировавший в конце января 2021 года, предназначен для снижения риска сердечно-сосудистой смерти и госпитализации по причине сердечной недостаточности у определенной категории пациентов.
Помимо «Веркуво» и «Винревэйр» (Winrevair, сотатерцепт) все остальные кардиометаболические лекарства «Мерк и Ко» являются экспериментальными. Так, потенциальный бестселлер энлицитид (enlicitide, MK-0616), пероральный ингибитор PCSK9, ориентирован на снижение холестерина липопротеинов низкой плотности (ЛПНП).
Антикоагулянт MK-2060, моноклонональное антитело против фактора XI (FXI), проверяется в задаче предотвращения тромбозов при терминальной стадии почечной недостаточности. Ингаляционный MK-5475, низкомолекулярный стимулятор sGC, изучается в лечении легочной артериальной гипертензии (ЛАГ) и ассоциированной с хронической обструктивной болезнью легких (ХОБЛ) легочной гипертензии. Эфинопегдутид (efinopegdutide, MK-6024, HM12525A, JNJ-64565111), лицензированный у корейской «Ханми фармасьютикал» (Hanmi Pharmaceutical), инъекционный двойной агонист GLP1R и GCGR, нацелен на лечение неалкогольного стеатогепатита (НАСГ).
«Мерк и Ко» (Merck & Co.) разработала энлицитида хлорид (enlicitide chloride, MK-0616) — новый лекарственный препарат, предназначенный для снижения уровня холестерина липопротеинов низкой плотности (ЛПНП) в целях лечения и профилактики атеросклероза.
Энлицитид обещает быть бестселлером. Во-первых, он безопасен. Во-вторых, лекарство сделано в удобной для применения пероральной рецептуре. В-третьих, энлицитид снижает уровень «плохого» холестерина существенно сильнее, чем все прочие существующие на рынке таблетки, включая секвестранты желчных кислот, статины, эзетемиб и бемпедоевую кислоту. В-четвертых, эффективность энлицитида не уступает таковой у мощных и дорогостоящих инъекционных препаратов для снижения уровня холестерина ЛПНП, таких как «Пралуэнт» (Praluent, алирокумаб), «Репата» (Repatha, эволокумаб) и «Леквио» / «Сибрава» (Leqvio / Sibrava, инклисиран).
Энлицитиду предстоит пройти масштабную клиническую проверку на сотнях и тысячах пациентов, которая позволит окончательно убедиться в безопасности и эффективности этого лекарственного средства.
Если всё окажется удачным, готовый препарат появится на рынке ориентировочно в 2026 году.
Энлицитид: механизм действия
Липопротеины низкой плотности (ЛПНП) — одна из пяти основных групп липопротеинов, которые переносят все молекулы жира по организму во внеклеточной воде. ЛПНП доставляют молекулы жира к клеткам и участвуют в развитии атеросклероза — процесса, при котором жир окисляется в стенках артерий [1].
Рецептор липопротеинов низкой плотности представляет собой мозаичный белок, опосредующий эндоцитоз ЛПНП, обогащенных холестерином. ЛПНП-рецепторы являются первичными рецепторами для транспорта ЛПНП из системного кровотока внутрь клеток: каждый переносит 3–6 тыс. молекул жира (включая холестерин) [2].
После того как ЛПНП-рецептор интернализирован, он диссоциирует от своего лиганда и возвращается на поверхность клетки. ЛПНП-рецептор совершает подобное путешествие в клетку и обратно каждые 10 минут, что составляет в общей сложности несколько сотен путешествий за 20 часов его жизни. Поскольку каждая частица ЛПНП содержит приблизительно 1600 молекул холестерина, подобная быстрая рециркуляция ЛПНП-рецепторов обеспечивает эффективный механизм доставки холестерина в клетки [3] [4] [5] [6].
Другими словами, ЛПНП-рецепторы поддерживают гомеостатический уровень ЛПНП в плазме крови. При этом печень отвечает за устранение приблизительно 70% циркулирующих ЛПНП. Чем обширнее доступный пул ЛПНП-рецепторов, тем эффективнее транспорт холестерина и ниже его концентрация в плазме крови.
Согласно липидной гипотезе (также известной как холестериновая гипотеза), снижение уровня холестерина в крови приводит к снижению риска сердечно-сосудистых заболеваний [7] [8] [9] [10] [11] [12].
Пропротеиновая конвертаза субтилизин-кексинового типа 9 (PCSK9) — один из важнейших регуляторов метаболизма холестерина ЛПНП. Этот фермент связывается с находящимися на поверхности гепатоцитов ЛПНП-рецепторами. В итоге последние вместо того, чтобы следовать своим физиологическим путем рециркуляторного возвращения к мембране гепатоцитов, перенаправляются к лизомальному разрушению в печени. Поскольку пул свободных ЛПНП-рецепторов сокращается, растет уровень холестерина ЛПНП в плазме [13] [14] [15].
PCSK9 также ингибирует внутриклеточное разрушение аполипопротеина B (ApoB) — белка, входящего в состав частиц ЛПНП и липопротеинов очень низкой плотности (ЛПОНП) [16] [17] [18]. Высокий уровень ApoB ассоциирован с высокой концентрацией частиц ЛПНП и является надежным индикатором сердечно-сосудистых заболеваний [19] [20] [21].
Терапевтическая блокада PCSK9 должна снизить плазматическую концентрацию ЛПНП-частиц, транспортирующих холестерин [22]. Опять же, согласно генетическим исследованиям, мутации PCSK9 с потерей функции приводят к пожизненно низкому уровню холестерина и заметному снижению риска атеросклеротического сердечно-сосудистого заболевания (АССЗ) [23] [24] [25] [26].
«Мерк и Ко» (Merck & Co.) задалась стратегической целью разработки перорального ингибитора PCSK9, который снижал бы холестерин ЛПНП на уровне, эквивалентном тому, который предоставляют одобренные моноклональные антитела — «Пралуэнт» (Praluent, алирокумаб) и «Репата» (Repatha, эволокумаб).
Химическая структура энлицитида (enlicitide). Изображение: C&EN.
Пероральный энлицитида хлорид (enlicitide chloride, MK-0616) представляет собой синтетический макроциклический (трициклический) пептид, который осуществляет ортостерическое блокирование белок-белкового взаимодействия между PCSK9 и рецептором ЛПНП. Энлицитид связывается с крупной плоской связывающей поверхностью на PCSK9, тем самым предотвращая связывание последнего с ЛПНП-рецептором. Итогом становится увеличение пула ЛПНП-рецепторов, доступных для клиренса холестерина ЛПНП [27] [28] [29].
Молекула характеризуется высокой потентностью связывания мишени: аффинность подобна на таковую у моноклональных антител против PCSK9. Желудочно-кишечная абсорбция энлицитида существенно улучшена путем использования усилителя проницаемости капрата натрия (sodium caprate).
Энлицитид разработан благодаря оформленному в апреле 2013 года партнерству между «Мерк и Ко» и «Ра фармасьютикалс» (Ra Pharmaceuticals), которая предоставила доступ к фирменной платформе цикломиметиков (cyclomimetic) и которую в апреле 2020 года купила бельгийская «ЮСиБи» (UCB).
Энлицитид: клиническая проверка
Начальные исследования
Первое клиническое испытание включило здоровых мужчин (n=60), рандомизированных для ежедневного получения энлицитида (enlicitide, MK-0616) в дозе от 10 до 300 мг или плацебо. Назначение экспериментального препарата привело к снижению уровня пропротеиновой конвертазы субтилизин-кексинового типа 9 (PCSK9) на более чем 90% от исходного, причем вне зависимости от дозы MK-0616.
Второе клиническое испытание привлекло мужчин и женщин (n=40) с высоким уровнем холестерина липопротеинов низкой плотности (ЛПНП), которые уже придерживались статиновой терапии. Участникам ежедневно назначали MK-0616 в дозе 10 или 20 мг либо плацебо. По прошествии 14 дней лечения энлицитид обеспечил снижение уровня холестерина ЛПНП на 65% от исходного. В группе плацебо снижение составило менее чем 5%.
Профиль безопасности энлицитида благоприятствовал: ежедневные дозы препарата вплоть до 300 мг не привели к каким-либо серьезным нежелательным явлениям.
Среднестадийное исследование
Клиническое испытание NCT05261126 фазы IIb (рандомизированное, двойное слепое, плацебо-контролируемое, многоцентровое, международное) пригласило взрослых пациентов (n=380) с гиперхолестеринемией.
[membership level=»2,3″ show_noaccess=»true»]
Среди основных характеристик участников: медиана возраста 62 года, 49% женщин; усредненный уровень холестерина липопротеинов низкой плотности (ЛПНП) составлял 119,5 мг/дл; атеросклеротическое сердечно-сосудистое заболевание (АСССЗ) в клинической форме у 39%, АСССЗ промежуточного или высокого риска у 56%, АСССЗ пограничного риска у 5%; не принимали статины, проходили статиновую терапию низкой или умеренной интенсивности, получали высокоинтенсивную терапию статинами соответственно 39%, 35% и 26%.
Испытуемым ежедневно назначали либо энлицитид (enlicitide, MK-0616) в дозе 6, 12, 18 или 30 мг, либо плацебо.
Первичная конечная точка эффективности лечения была установлена процентным изменением уровня холестерина ЛПНП.
По прошествии 8 недель в 6-, 12-, 18 и 30-мг подгруппах энлицитида было зарегистрированоснижение уровня холестерина ЛПНП на 40%, 55%, 58% и 60% — против его роста на 1,2% в группе плацебо.
Соответствующая разница, скорректированная на плацебо, получилась статистически значимой (p<0,001) и равной −41%, −56%, −59% и −61%.
К целевому уровню холестерина ЛПНП, рассчитываемому в зависимости от степени риска АСССЗ, вышли 81%, 86%, 91% и 91% пациентов — против 9% в группе контроля.
Среди прочих благотворных эффектов, оказанных энлицитидом в ходе лечения гиперхолестеринемии и отраженных изменением относительно плацебо уровней других метаболических показателей, выступающих важными предикторами атерогенного риска:
холестерин липопротеинов высокой плотности (ЛПВП): −2%, +2%, +4%, −1%;
общий холестерин: −27%, −37%, −39%, −42%;
триглицериды: −8%, −14%, −16%, −16%;
холестерин липопротеинов очень низкой плотности (ЛПОНП): −6%, −14%, −14%, −13%.
Применение энлицитида характеризовалось приемлемой переносимостью. Частоты нежелательных явлений (НЯ) оказались идентичными в подгруппах препарата и контрольной группе: 40–44% против 44%.
Среди наиболее распространенных НЯ, протекавших главным образом с легкой степенью выраженности: коронавирусная инфекция COVID-19 (у 3–8% пациентов, получавших энлицитид, — против 9% в группе плацебо), диарея (1–7% против 9%), диспепсия (0–7% против 1%), усталость (0–4% против 2%), артралгия (0–5% против 2%), тошнота (1–5% против 0%).
Не зафиксировано НЯ, которые проявились бы дозозависимым образом в результате лечения. Из-за НЯ прием энлицитида прекратили 2% пациентов — против 1% в группе плацебо.
[/membership]
Позднестадийные исследования
В конце августа 2023 года «Мерк и Ко» (Merck & Co.) объявила о запуске опорной клинической программы CORALreef, которая охватит приблизительно 17 тыс. человек и проводимые в рамках которой испытания фазы III лягут в основу регистрационного досье энлицитида (enlicitide, MK-0616), предназначенного для снижения уровня холестерина липопротеинов низкой плотности (ЛПНП):
[membership level=»2,3″ show_noaccess=»true»]
CORALreef Lipids (NCT05952856): взрослые пациенты (n=2760) с гиперхолестеринемией, у которых в анамнезе либо уже было серьезное осложнение атеросклеротического сердечно-сосудистого заболевания (АССЗ) и уровень холестерина ЛПНП ≥ 55 мг/дл, либо такого осложнения еще не было, но присутствует умеренно-высокий риск его развития, а уровень холестерина ЛПНП ≥ 70 мг/дл. Под серьезным осложнением АССЗ понимают острый коронарный синдром, инфаркт миокарда, ишемический инсульт, симптоматическая болезнь периферических артерий. Первичная конечная точка эффективности лечения — усредненное процентное изменение уровня холестерина ЛПНП после 24 недель терапии.
CORALreef HeFH (NCT05952869): взрослые пациенты (n=270) с гетерозиготной семейной гиперхолестеринемией, уровнем холестерина ЛПНП ≥ 55 мг/дл или ≥ 70 мг/дл (в зависимости от истории болезни), придерживающиеся статиновой терапии умеренно-высокой интенсивности. Первичная конечная точка эффективности лечения — усредненное процентное изменение уровня холестерина ЛПНП после 24 недель терапии.
CORALreef Outcomes (NCT06008756): взрослые пациенты (n=14550), придерживающиеся статиновой терапии умеренно-высокой интенсивности и с уровнем холестерина ЛПНП ≥ 70 мг/дл или ≥ 90 мг/дл (в зависимости от истории болезни), либо с серьезным осложнением АССЗ в анамнезе (инфаркт миокарда, ишемический инсульт, успешная реваскуляризация периферических артерий, большая ампутация по причине атеросклероза), либо с высоким риском развития первого серьезного осложнения АССЗ (возраст ≥ 50 лет и признаки болезни коронарных артерий, болезни периферических артерий или атеросклеротического цереброваскулярного заболевания; возраст ≥ 60 лет и сахарный диабет, а также микрососудистое заболевание, соотношение альбумина к креатинину в моче ≥ 40 мг/ммоль, ежедневная инсулинотерапия или диабет продолжительностью ≥ 10 лет). Первичная конечная точка эффективности лечения — время до наступления первого серьезного сердечно-сосудистого события (MACE), включая болезнь коронарных артерий со смертельным исходом, инфаркт миокарда, летальный и нелетальный ишемический инсульт, острая ишемия конечностей или большая ампутация, срочная артериальная реваскуляризация (коронарная, цереброваскулярная, периферическая).
Результаты клинических исследований CORALreef Lipids и CORALreef HeFH будут готовы к сентябрю 2025 года, тогда как итоги CORALreef Outcomes будут подведены только осенью 2029-го, что объясняется желанием выяснить долгосрочные шестилетние исходы при постоянном назначении энлицитида.
[/membership]
Экспертные комментарии
Терапия пероральными статинами давно является золотым стандартом в лечении гиперхолестеринемии [1] [2]. Однако, несмотря на руководящие принципы, в которых основное внимание уделено использованию высокоинтенсивной статиновой терапии, применение препаратов этого класса в клинической практике остается сложной задачей, что может быть частично связано с нежелательными явлениями и непереносимостью статинов. Кроме того, даже при успешном внедрении статины в высоких дозах часто не способствуют достижению целей лечения, особенно у пациентов с высоким риском [3] [4] [5].
[membership level=»2,3″ show_noaccess=»true»]
Добавление нестатиновых препаратов с альтернативными механизмами действия является рекомендованной стратегией, но многие пациенты всё равно остаются недолеченными [6] [7] [8] [9] [10]. Эффективность снижения уровня холестерина липопротеинов низкой плотности (ЛПНП) при помощи дополнительных пероральных лекарственных средств относительно скромная. Так, секвестранты желчных кислот добавляют к снижению уровня холестерина ЛПНП приблизительно 12–18%, эзетемиб (ezetimibe) — 20–25%, бемпедоевая кислота (bempedoic acid) — 18% [2] [11] [12].
Экспериментальный энлицитид (enlicitide, MK-0616), разрабатываемый «Мерк и Ко» (Merck & Co.) пероральный ингибитор пропротеиновой конвертазы субтилизин-кексинового типа 9 (PCSK9), обеспечил, в отличие от вышеуказанных пероральных препаратов, существенно более глубокое снижение уровня холестерина ЛПНП — на 41–61% с поправкой на плацебо.
Класс лекарственных средств, таргетированных против PCSK9, существует давно. Его первыми представителями стали моноклональные антитела «Пралуэнт» (Praluent, алирокумаб) и «Репата» (Repatha, эволокумаб), предложенные соответственно «Санофи» (Sanofi) / «Ридженерон фармасьютикалс» (Regeneron Pharmaceuticals) и «Амджен» (Amgen) летом 2015 года.
Алирокумабу (alirocumab) и эволокумабу (evolocumab) по силам снижать уровень холестерина ЛПНП в среднем на 48–56% относительно плацебо [13] [14]. Несмотря на редкое и удобное для пациента дозирование, реализованное подкожными инъекциями один раз в месяц или каждые две недели, большим спросом «Пралуэнт» и «Репата» не пользуются ввиду их высокой стоимости, покрывать которую поставщики страховых медицинских услуг особым желанием не горят.
И правда: если курс лечения статинами, много лет назад перешедшими в разряд генерических лекарств, обходится в сущие копейки, то за годовой курс лечения «Пралуэнтом» или «Репатой» требовалось заплатить 14 тыс. долларов с гаком. Впрочем, через которое время, когда производители осознали, что спрос катастрофически низок, ценник был резко сброшен на 60% — до без малого 6 тыс. долларов.
В конце декабря 2020 года «Новартис» (Novartis) выпустила «Леквио» (Leqvio, инклисиран) по американской цене 6,5 тыс. долларов в год. Ключевой особенностью инклисирана (inclisiran), построенного на базе РНК-интерференции, является режим дозирования: в поддерживающем режиме он применяется подкожно один раз в шесть месяцев. Терапевтическая эффективность «Леквио» находится в пределах 48–52-процентного снижения уровня холестерина ЛПНП относительно плацебо [15].
Две инъекции инклисирана в год — и о высоком холестерине можно забыть.
Механистическое отличие указанной тройки препаратов состоит в том, что алирокумаб и эволокумаб блокируют циркулирующую PCSK9, тогда как инклисиран подавляет непосредственно синтез PCSK9.
Учитывая высококонкурентную обстановку на рынке PCSK9-ингибиторов, нет никаких сомнений, что привлечь внимание пациентского, врачебного и страхового сообщества к энлицитиду можно лишь одним — ценовой доступностью.
Что говорить, если инклисиран, в середине апреля 2022 года получивший регистрацию в России под брендовым названием «Сибрава» (Sibrava), стоит дорого: от 80 до 130 тыс. рублей за одну инъекцию, то есть 160–260 тыс. рублей в год. В перечень жизненно необходимых и важнейших лекарственных препаратов (ЖНВЛП) для медицинского применения инклисиран не вошел ни в 2023 году, ни в 2024-м.
Стоимость лечения гиперхолестеринемии при помощи «Пралуэнта» или «Репаты» доходит в России до запредельных значений. Так, годовой курс алирокумаба или эволокумаба встанет в сумму, приблизительно равную 415–460 тыс. рублей в год. И это в лучшем случае, ведь для некоторых пациентов, требующих более интенсивной терапии, лечение может обойти существенно дороже: от 920 тыс. до 1,24 млн рублей в год. Оба препарата включены в список ЖНВЛП.
Инъекционная природа указанной тройки медикаментов против «плохого» холестерина — «Пралуэнта», «Репаты», «Леквио» / «Сибравы», пусть даже вводимых не внутривенно, а подкожно в домашних условиях, всё же не добавляет им притягательности с точки зрения пациентов: куда проще принять пероральный статин и считать, пусть даже совершенно ошибочно, что всё идеально и в порядке.
Многообещающая терапевтическая эффективность энлицитида наряду с его безопасностью и удобством таблеточного применения должны найти подкрепление в продолжительных клинических испытаниях. Короткой 8-недельной проверки никак недостаточно для получения убедительных доказательств, особенно среди различных популяций пациентов.
Если энлицитид будет одобрен, что может произойти ближе к 2026 году, борьба с атеросклерозом, которым страдает половина взрослого населения планеты, выйдет на качественно новый уровень — благодаря эффективности и удобству грядущего медикамента.
Нынешняя ситуация с лечением сердечно-сосудистых заболеваний удручает: до половины пациентов прекращают прием лекарственных препаратов в течение года после начала терапии [16] [17] и столько же людей, которым показаны статины, не принимают их [18], притом что лишь 2% больных, которым рекомендованы PCSK9-ингибиторы, ими лечатся [19].
Можно смело утверждать, что по стоимости энлицитиду никак не получится стать доступным каждому, как это наблюдается с дешевыми статинами. Следует рассчитывать, что цена энлицитида будет не ниже таковой у бемпедоевой кислоты: в США стоимость годового курса препаратом «Некслетол» (Nexletol) составляет 5340 долларов для пациентов без страхового покрытия. Всё зависит от «Мерк и Ко», которая легко может пойти на поводу алчности своих акционеров, запросив за энлицитид много денег, тем самым по сути сведя в могилу бизнес-перспективы этого препарата. Показателен пример «Эсперион терапьютикс» (Esperion Therapeutics), которая заработала на бемпедоевой кислоте скромные суммы в 75,5 и 78,4 млн долларов в 2022 и 2021 гг.
Согласно отраслевым прогнозам, энлицитид на пике продаж способен добраться до 5 млрд долларов. И это весьма оптимистично, учитывая, что весь присутствующий на мировом рынке класс PCSK9-ингибиторов наторговал на 2,3 и 3,1 млрд долларов в 2022 и 2023 гг.
[/membership]
Экспериментальный конвейер PCSK9-ингибиторов
Ряд игроков фармотрасли трудятся над новыми ингибиторами пропротеиновой конвертазы субтилизин-кексинового типа 9 (PCSK9), предназначенными для профилактики атеросклеротических сердечно-сосудистых осложнений при дислипидемии — путем снижения уровня холестерина липопротеинов низкой плотности (ЛПНП).
Пероральные препараты
«АстраЗенека» (AstraZeneca) — единственная, кто активно разрабатывает пероральный PCSK9-ингибитор, являющийся прямым конкурентом энлицитида (enlicitide, MK-0616) авторства «Мерк и Ко» (Merck & Co.).
[membership level=»2,3″ show_noaccess=»true»]
AZD0780 был куплен у «Догма терапьютикс» (Dogma Therapeutics) в середине сентября 2020 года. Клиническое испытание PURSUIT (NCT06173570) фазы IIb, в котором экспериментальный препарат назначается ежедневно в четырех разных дозах пациентам с дислипидемией, завершится к концу лета 2024 года.
AZD0780 представляет собой антисмысловой олигонуклеотид (ASO), который связывает комплементарную мРНК гена PCSK9, чтобы заглушить его экспрессию с результирующим подавлением трансляции одноименного белка. Пероральная доставка ASO сопряжена с рядом трудностей, поскольку эти соединения гидрофильны, должны проходить через кислую среду желудка и плохо всасываются в кишечнике. Благодаря модификации ASO капратом натрия (sodium caprate) для повышения кишечной проницаемости и кишечнорастворимой оболочке для защиты от агрессивной кислотной среды желудка проблемы удалось обойти: доклиническая проверка подтвердила клинически значимую биодоступность AZD0780 в печени [1] [2].
Приблизительно этим же занималась «Ново Нордиск» (Novo Nordisk), но в ноябре 2022 года программа перорального PCSK9-ингибитора NNC0385-0434 (NN6435) была свернута «ввиду коммерческих и портфельных соображений».
NNC0385-0434, будучи низкомолекулярным пептидным ингибитором PCSK9, располагает химической структурой, схожей с рецептором ЛПНП: используется EGF-подобный домен A последнего. И потому молекула конкурентно связывает свободный PCSK9, тем самым как бы оттягивая его внимание от связывания с естественным ЛПНП-рецептором [3].
Вопрос кардинального улучшения пероральной биодоступности NNC0385-0434 был решен добавлением каприлата натрия (caprylate sodium, SNAC) — такого же усилителя всасываемости, который используется в рецептуре перорального семаглутида (semaglutide), агониста рецептора глюкагоноподобного пептида 1 (GLP1R) [4].
В клиническом исследовании NCT04992065 фазы II среди пациентов с атеросклеротическим сердечно-сосудистым заболеванием (АССЗ) или риском его развития ежедневное назначение NNC0385-0434 в дозе 15, 40 или 100 мг привело к снижению уровня холестерина ЛПНП на 32%, 45% и 62% относительно плацебо [3].
Что примечательно, группа активного сравнения, получавшая моноклональное антиа-PCSK9-нтитело «Репата» (Repatha, эволокумаб) подкожными инъекциями каждые две недели, продемонстрировала снижение исходного уровня холестерина ЛПНП на 59,6%, тогда как 100-мг доза NNC0385-0434 обеспечила снижение на 56,2%, то есть разойдясь всего лишь на 3,4%.
Ничего конкретного не слышно об антисмысловом олигонуклеотиде цепадакурсен (cepadacursen, CIVI 007), который открыла датская «Сантарис фарма» (Santaris Pharma), впоследствие поглощенная «Рош» (Roche), и который «Сиви байофарма» (CiVi Biopharma) планирует превратить в пероральное лекарство CIVI 008. Во всяком случае, на четвертый квартал 2023 года была запланирована отправка в Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) заявки на проведение клинического испытания фазы I/IIa.
Для реализации пероральной доставки CIVI 008 используется носитель в лице соли N-(5-хлорсалицилоил)-8-аминокаприловой кислоты (5-CNAC).
Как предполагает «Сиви», стоимость годового курса CIVI 008 для американских пациентов будет находиться в пределах 2–3 тыс. долларов.
Результаты клинических исследований NCT03427710 фазы I и NCT04164888 фазы IIa цепадакурсена среди пациентов с гиперхолестеринемией не опубликованы.
Китайская «Си-ви-ай фармасьютикалс» (CVI Pharmaceuticals) придумала CVI-LM001, пероральный низкомолекулярный PCSK9-модулятор, который повышает экспрессию ЛПНП-рецепторов в печени и ускоряет выведение холестерина ЛПНП из циркуляции посредством двойного механизма действия: препарат ингибирует транскрипцию PCSK9 и предотвращает деградацию мРНК рецептора ЛПНП.
В клиническом исследовании фазы Ib ежедневный прием CVI-LM001 пациентами с гиперлипидемией обеспечил снижение уровня холестерина ЛПНП на 26% относительно плацебо [5]. Впрочем, дальнейшая разработка застопорилась: клиническое испытание NCT04438096 фазы II, начатое в середине августа 2020 года, неактивно.
Китайская «Гуанчжоу Цзоё фарма» (Guangzhou Joyo Pharma) пробует силы с DC371739 — пероральным низкомолекулярным соединением, которое напрямую связывает транскрипционный фактор HNF1A, тем самым препятствуя транскрипции генов PCSK9 и ангиопоэтин-подобного белка 3 (ANGPTL3) [6]. Последний, будучи ингибитором липопротеинлипазы (LPL) и эндотелиальной липазы (LIPG), занимает определяющее место в метаболизме липопротеинов, притом что он модулирует холестерин ЛПНП путем, который не зависит от ЛПНП-рецептора и других известных механизмов, ответственных за клиренс холестерина ЛПНП из плазмы.
В начале февраля 2021 года появился ANGPTL3-ингибитор «Эвкиза» (Evkeeza, эвинакумаб), который «Ридженерон фармасьютикалс» (Regeneron Pharmaceuticals) предлагает для лечения гомозиготной семейной гиперхолестеринемии.
Эвинакумаб поможет снизить уровень холестерина, когда бессильны все прочие липидоснижающие препараты.
Разработка DC371739 отталкивалась от молекулярной конструкции тетрагидропротоберберинов, извлеченных из хохлатки обманчивой (Corydalis ambigua) — растения, применяемого в китайской народной медицине.
В клиническом исследовании NCT04927221 фазы I среди пациентов с гиперхолестеринемией ежедневный прием DC371739 снизил уровень холестерина ЛПНП на 19% относительно плацебо [6].
[/membership]
Антисмысловые олигонуклеотиды
В конце сентября 2022 года «АстраЗенека» (AstraZeneca)отказалась от совместного с «Айонис фармасьютикалс» (Ionis Pharmaceuticals) антисмыслового олигонуклеотида AZD8233 (ION449) против PCSK9, назначаемого ежемесячными подкожными инъекциями. Оригинатор в лице «Айонис» самостоятельно продолжать дорогостоящее развитие препарата не заинтересован.
[membership level=»2,3″ show_noaccess=»true»]
Сворачивание программы связано с нагнетаемой конкурентной обстановкой. Во-первых, уже есть моноклональные антитела, которые по аналогии применяются подкожно один раз в месяц и которые обеспечивают приличную эффективность. Во-вторых, неминуемо появление перорального энлицитида.
В клиническом исследовании SOLANO (NCT04964557) фазы IIb среди пациентов с гиперлипидемией AZD8233 в дозе 60 мг обеспечил снижение уровня холестерина ЛПНП на 62% относительно плацебо.
В предшествовавшем клиническом исследовании ETESIAN (NCT04641299) фазы IIb среди пациентов с гиперхолестеринемией и высоким сердечно-сосудистым риском назначение AZD8233 в дозе 50 или 90 мг привело к снижению уровня холестерина ЛПНП на 71% и 77% относительно плацебо [1].
[/membership]
Моноклональные антитела
В середине августа 2023 года китайская «Инновент байолоджикс» (Innovent Biologics) получила одобрение местного регулятора на PCSK9-ингибитор «Синтбило» (Sintbilo, тафолецимаб). Моноклональное антитело тафолецимаб (tafolecimab, IBI306), подкожно назначаемое каждые 4 или 6 недель пациентам с первичной гиперхолестеринемией (включая несемейную и гетерозиготную семейную) и смешанной дислипидемией, обеспечило снижение уровня холестерина ЛПНП в среднем на 61% относительно плацебо.
[membership level=»2,3″ show_noaccess=»true»]
Другие китайские фармразработчики готовятся предложить свои подкожные PCSK9-ингибиторы: моноклональные антитела эбронуцимаб (ebronucimab, AK102), рекатицимаб (recaticimab, SHR-1209) и онгерицимаб (ongericimab, JS002) разработаны соответственно «Акесо» (Akeso), «Цзянсу Хэнжуй фармасьютикалс» (Jiangsu Hengrui Pharmaceuticals), «Шанхай Цзюньши байосайенсиз» (Shanghai Junshi Biosciences).
Интерес представляет рекатицимаб, который в режиме применения каждые 2 или 3 месяца, снизил уровень холестерина ЛПНП на 53% и 45% относительно плацебо.
[/membership]
Вакцины
Биотехнологическая экспертиза «Ваксинити» (Vaxxinity), входящей в состав «Юнайтед байомедикал» (United Biomedical), построена вокруг синтетических пептидных вакцин, стимулирующих выработку эндогенных антител против заданных мишеней.
[membership level=»2,3″ show_noaccess=»true»]
Оптимальное сочетание фирменных синтетических T-хелперных пептидов UBITh, прилинкованных к таргетным эпитопам, определяет избирательную активацию иммунной системы, преодолевает иммунную толерантность к эндогенным белкам в целях индуцирования целевого гуморального B-клеточного ответа, минимизирует риски иммунного уклонения и опосредованной T-клетками цитотоксичности.
Доклиническая проверка вакцинного кандидата VXX-401 на нечеловекообразных приматах продемонстрировала устойчивое подавление PCSK9, что отразилось снижением холестерина ЛПНП на 30–50%. Иммунизация переносилась хорошо и подтвердила факт преодоления иммунной толерантности [1] [2].
Продолжается клиническое исследование NCT05762276 фазы I, которое оценивает безопасность, переносимость, иммуногенность и фармакодинамику VXX-401 среди здоровых добровольцев, а также выясняет оптимальную схему внутримышечного дозирования этого вакцинного препарата-кандидата. Результаты появятся к середине 2024 года.
Австрийская «Аффирис» (Affiris) занимается специфической активной иммунотерапией (SAIT), целью которой является длительное и устойчивое подавление патогенных белков-мишеней путем активации иммунной системы организма, чтобы заставить ее вырабатывать соответствующие антитела. После примирования терапевтической вакциной последующая бустерная иммунизация назначается редко: раз в год, полгода или квартал.
Ноу-хау «Аффирис» построено вокруг биотехнологической платформы AFFITOME. Создаются аминокислотные последовательности (в виде коротких пептидов), имитирующие участки эпитопов белка-мишени в организме. Далее они посредством мутагенеза модифицируются так, чтобы быть максимально «чужеродными» для иммунной системы (для усиления иммунного ответа). Затем эти иммуногенные пептидные «двойники», названные аффитопами (affitope), объединяются с белковым носителем (для доставки в организм) и адъювантным гидроксидом алюминия (для усиления иммунного ответа). После подкожного введения аффитопы распознаются иммунной системой, которая начинает вырабатывать против них антитела, которые параллельно атакуют патогенные белки-мишени.
Вакцинный кандидат AT04 (FB6001), содержащий 10 аминокислотных последовательностей и разработанный для разрушения иммунной толерантности к белку PCSK9, индуцирует специфический ответ олигоклональных антител, которые перекрестно реагируют и ингибируют белок-мишень в лице PCSK9, причем без активации аутоиммунитета.
В ходе клинического исследования NCT02508896 фазы I здоровые добровольцы прошли примирование тремя дозами терапевтической вакцины AT04 (на 0-й, 4-й и 8-й неделе), а затем получили одну бустерную дозу (на 60-й неделе, то есть спустя год). Снижение уровня холестерина ЛПНП относительно плацебо составило 11% и 13% на 20-й и 70-й неделе. На протяжении всего испытания усредненное снижение вышло к 7% относительно плацебо, максимально зарегистрированное — 39% [3].
В конце декабря 2021 года китайская «Фронтир байотекнолоджис» (Frontier Biotechnologies) лицензировала AT04 для лечения гиперхолестеринемии.
Нет сведений, продолжает ли кто-нибудь разработку AT04.
[/membership]
Генное редактирование
Не исключено, в долгосрочной перспективе сектор PCSK9-ингибиторов изменится кардинальным образом, когда станет доступна терапевтическая модальность, предполагающая однократное лечение. Так, VERVE-101 авторства «Верв терапьютикс» (Verve Therapeutics) представляет собой генное редактирование in vivo, когда доставляемые в гепатоциты мРНК-инструкции меняют, обращаясь к технологии CRISPR, одну пару ДНК-оснований гена, кодирующего PCSK9, тем самым нарушая производство соответствующего белка.
[membership level=»2,3″ show_noaccess=»true»]
В доклиническом исследовании на нечеловекообразных приматах одна внутривенная инфузия экспериментального препарата VERVE-101 обеспечила отключение PCSK9-гена с 70-процентной эффективность, что отразилось снижением уровня PCSK9-белка в крови на 89% и снижением уровня холестерина ЛПНП на 69%. Результаты сохранялись на всём протяжении наблюдений сроком 1,3 года. Что важно, не выявлено предпосылок для передачи отредактированного PCSK9-гена потомству [1].
Продолжается клиническое исследование HEART-1 (NCT05398029) фазы Ib, тестирующее нокдаун гена PCSK9 среди пациентов с гетерозиготной семейной гиперхолестеринемией с развившимися атеросклеротическим сердечно-сосудистым заболеванием (АССЗ) и неконтролируемой гиперхолестеринемией.
Согласно промежуточному анализу данных трех пациентов, собранных по прошествии одного месяца после введения VERVE-101, два испытуемых из группы низкой дозы продемонстрировали снижение уровня PCSK9-белка на 59% и 84% при сопутствующем снижении уровня холестерина ЛПНП на 39% и 48%. Один человек из группы высокой дозы показал соответствующие снижения на 47% и 55%, притом что достигнутое падение уровня холестерина ЛПНП сохранялось на протяжении 180 дней.
В первой половине 2024 года «Верв» планирует запуск клинического исследования фазы Ib конструктивно усовершенствованного генного редактирования VERVE-102, которое, как предполагается, повысит эффективность отключения гена PCSK9.
Усилия «Верв» поддерживаются со стороны «Илай Лилли» (Eli Lilly).
«Присижн байосайенсиз» (Precision Biosciences) подтвердила, что целевой эффект снижения уровня холестерина ЛПНП, осуществленный у нечеловекообразных приматов посредством генного вмешательства in vivo в структуру гена PCSK9, сохранился на протяжении 3 лет наблюдений [2].
Генное редактирование авторства «Присижн» реализуется посредством фирменной платформы ARCUS, использующей I-CreI — естественный фермент редактирования генома, обнаруженный в водоросли Chlamydomonas reinhardtii и предназначенный для высокоспецифичных разрезов и вставок в клеточную ДНК. I-CreI относится к более крупному классу ферментов, называемых самонаводящимися эндонуклеазами или мегануклеазами.
Впрочем, в начале января 2023 года «Присижн» поставила на паузу программу PBGENE-PCSK9 ввиду трудностей с получением разрешения FDA на проведение клинических испытаний.
[/membership]
Прочие
В первой половине 2024 года «Эл-ай-би терапьютикс» (LIB Therapeutics) отправит на регистрацию леродалцибеп (lerodalcibep, LIB003) — малый рекомбинантный слитый белок из PCSK9-связывающего домена (аднектина) и человеческого сывороточного альбумина. Леродалцибеп, стабильный при комнатной температуре и подкожно вводимый раз в месяц, привел к снижению уровня холестерина ЛПНП на 59% относительно плацебо [1].
«Мерк и Ко» (Merck & Co.) уведомила об успешной опорной клинической проверке адъювантного назначения «Китруды» (Keytruda, пембролизумаб) пациентам, прошедшим хирургическую резекцию локализованной мышечно-инвазивной уротелиальной карциномы или местнораспространенной уротелиальной карциномы.
Применение пембролизумаба (pembrolizumab), блокатора PD-1, оказалось эффективнее, чем просто наблюдение, в задаче сдерживания рецидива заболевания.
В конце августа 2021 года «Опдиво» (Opdivo, ниволумаб), блокатор PD-1 авторства «Бристол-Майерс Сквибб» (Bristol-Myers Squibb), получил одобрение Управления по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) для адъювантного лечения уротелиальной карциномы у взрослых пациентов в группе высокого риска рецидива заболевания после радикальной резекции.
Комбинация из энфортумаба ведотина и пембролизумаба продлит жизнь при неоперабельной уротелиальной карциноме.
Клинические подробности
Клиническое исследование AMBASSADOR/KEYNOTE-123 (NCT03244384) фазы III (рандомизированное, открытое, многоцентровое, международное) пригласило взрослых пациентов (n=702), которые прошли хирургическую резекцию локализованной мышечно-инвазивной уротелиальной карциномы и местнораспространенной уротелиальной карциномы.
Участникам либо назначали пембролизумаб (pembrolizumab) каждые три недели сроком до 18 циклов (до момента прогрессирования заболевания или неприемлемой токсичности), либо за ними активно наблюдали.
Первичные конечные точки эффективности адъювантной терапии были установлены общей выживаемостью (OS) и безрецидивной выживаемостью (DFS).
Согласно промежуточному анализу собранных данных, после наблюдений в течение медианных 22,3 месяца применение «Китруды» обеспечило статистически и клинически значимое продление DFS: медиана этого показателя вышла к 29,0 месяца (95% ДИ [здесь и далее]: 21,8–NE) — против 14,0 месяца в группе контроля.
Снижение риска рецидива заболевания или смерти составило 31%: отношение риска (hazard ratio, HR) 0,69 (0,54–0,87; p=0,001).
Продление DFS оказалось справедливым вне зависимости от статуса экспрессии PD-L1.
На данном этапе пембролизумаб не смог вывести OS к статистически значимому расхождению с группой сравнения: после наблюдений на протяжении медианных 36,9 месяца медиана OS получилась равной 50,9 месяца (43,8–NE) — против 55,8 месяца (53,3–NE). Риск смерти снизился на 2%: HR 0,98 (0,76–1,26; p=0,88.
Экспертные комментарии
Радикальная хирургическая операция, включающая цистэктомию при опухолях мочевого пузыря или нефроуретерэктомию при опухолях верхних мочевых путей, является стандартом лечения пациентов с мышечно-инвазивной уротелиальной карциномой [1] [2].
Несмотря на то что хирургическое вмешательство осуществляется в целях полного излечения, более половины пациентов с патологическими признаками инвазии рака через мышечную оболочку или с вовлечением регионарных лимфатических узлов сталкиваются с летальным метастатическим рецидивом [1] [2] [3] [4].
Адъювантная химиотерапия продлевает безрецидивную выживаемость пациентов с местнораспространенной уротелиальной карциномой верхних путей [5], однако консенсуса относительно рутинной адъювантной химиотерапии цисплатином не выработано, а некоторые пациенты с уротелиальной карциномой не подходят или отказываются от неоадъювантной химиотерапии цисплатином [1] [2] [6] [7] [8] [9].
Опять же, несмотря на высокий риск метастатического рецидива, не было продемонстрировано, что стандартная адъювантная системная терапия улучшает исходы у пациентов с патологическими признаками резидуальной (остаточной) болезни после неоадъювантной химиотерапии цисплатином [10] [11] [12].
Надофараген фираденовек позволит избежать радикальной цистэктомии.
«Бристол-Майерс Сквибб» (Bristol-Myers Squibb) решила исправить ситуацию, и по итогам ее PD-1-блокатор «Опдиво» (Opdivo, ниволумаб) в конце августа 2021 года был одобрен Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) для адъювантного лечения уротелиальной карциномы у взрослых пациентов в группе высокого риска рецидива заболевания после радикальной резекции.
«Мерк и Ко» (Merck & Co.) для усиления позиций «Китруды» (Keytruda, пембролизумаб) в противостоянии с «Опдиво» необходимо обойти те результаты, которые сгенерировал последний. Если согласовать периоды наблюдений, то клинические исходы, похоже, в целом не отличаются. Во всяком случае продемонстрировано, что и пембролизумаб (pembrolizumab), ниволумаб (nivolumab) приблизительно вдвое продлевают выживаемость без рецидива. В популяции пациентов с наличием опухолевой экспрессии PD-L1 (≥ 1%) улучшения оказались лучше, чем без данного биомаркера.
При этом, однако, следует отметить явную проблему с тем, что подразумевается под положительной экспрессией PD-L1. Разработчики лекарств по-разному подходят к этой оценке. Так, «Мерк и Ко» определяет уровень экспрессии PD-L1 по комбинированному показателю позитивности (CPS), который отталкивается от PD-L1-экспрессии на опухолевых и инфильтрирующих иммунных клетках (включая лимфоциты и макрофаги). «Бристол-Майерс Сквибб» исходит из PD-L1-экспрессии только на опухолевых клетках (TC). «АстраЗенека» (AstraZeneca) со своим PD-L1-блокатором «Имфинзи» (Imfinzi, дурвалумаб) обращается к иммуногистохимической PD-L1-экспрессии на инфильтрирующих опухоль иммунных клетках (TIIC).
Таким образом, ассоциации, наблюдаемые между экспрессией PD-L1 и клиническими исходами, различаются в исследованиях блокаторов PD-(L)1, проводимых разными фармкомпаниями. К примеру, в ходе лечения одинакового онкологического заболевания может получаться так, что при назначении дурвалумаба (durvalumab) повышенный уровень экспрессии PD-L1 коррелирует с углубленным ответом на терапию, тогда как при применении пембролизумаба или ниволумаба их клиническая активность не зависит от этого уровня.
Регистрационное клиническое исследование CheckMate 274 (NCT02632409) фазы III (рандомизированное, двойное слепое, плацебо-контролируемое, многоцентровое, международное) установило, что адъювантное назначение ниволумаба обеспечило следующие исходы, зафиксированные по прошествии наблюдений в течение медианных 20,9 месяца (0,1–48,3) и 19,5 месяца (0,0–50,0) в группе «Опдиво» и плацебо соответственно:
медиана безрецидивной выживаемости (DFS): 20,8 месяца (95% ДИ [здесь и далее]: 16,5–27,6) — против 10,8 месяца (8,3–13,9) в группе плацебо, отношение риска (hazard ratio, HR) 0,70 (98,22% ДИ: 0,55–0,90; p<0,001);
медиана DFS при экспрессии PD-L1 ≥ 1%: HR 0,56 (0,40–0,80);
медиана DFS при экспрессии PD-L1 < 1%: HR 0,82 (0,63–1,06).
медиана выживаемости без рецидивов за пределами уротелиального тракта (NUTRFS; без рецидивов в мягких тканях таза или с вовлечением тазовых узлов ниже бифуркации аорты): 22,9 месяца (19,2–33,4) — против 13,7 месяца (8,4–20,3), HR 0,72 (0,59–0,89);
медиана выживаемости без отдаленных метастазов (DMFS): 40,5 месяца (22,4–NE) — против 29,5 месяца (16,7–NE), HR 0,75 (0,59–0,94).
Дальнейшие наблюдения в течение 3 лет (медианных 36,1 месяца) зарегистрировалиулучшение показателей в группе «Опдиво»:
медиана DFS: 25,6 месяца (19,2–41,8) — против 8,5 месяца (7,3–13,7), HR 0,63 (0,51–0,78);
медиана DFS при экспрессии PD-L1 ≥ 1%: 52,6 месяца (39,5–NE) — против 8,3 месяца (4,7–15,2), HR 0,44 (0,30–0,63);
медиана DFS при экспрессии PD-L1 < 1%: 18,3 месяца (14,1–22,4) — против 9,7 месяца (7,4–16,6), HR 0,74 (0,56–0,97).
медиана NUTRFS: 25,8 месяца (19,2–44,0) — против 9,6 месяца (7,6–13,8), HR 0,64 (0,52–0,80);
медиана NUTRFS при экспрессии PD-L1 ≥ 1%: 52,6 месяца — против 8,4 месяца, HR 0,53 (0,38–0,74).
медиана DMFS: 41,8 месяца (25,6–55,3) — против 19,4 месяца (11,4–34,4), HR 0,70 (0,55–0,89);
медиана DMFS при экспрессии PD-L1 ≥ 1%: NR — против 20,7 месяца, HR 0,58 (0,40–0,84).
медиана времени от рандомизации до прогрессирования заболевания после системной противоопухолевой терапии следующей линии, начала второй системной противоопухолевой терапии следующей линии или смертельного исхода (PFS2): 61,2 месяца (44,0–NE) — против 47,1 месяца (29,4–65,2), HR 0,79 (0,63–0,98);
медиана общей выживаемости (OS) еще не созрела.
Результаты в популяции пациентов с мышечно-инвазивным раком мочевого пузыряследующие:
медиана DFS: 25,6 месяца (19,2–41,8) — против 8,5 месяца (7,3–13,7), HR 0,63 (0,51–0,78);
медиана NUTRFS: 25,8 месяца (19,2–44,0) — против 9,6 месяца (7,6–13,8), HR 0,64 (0,52–0,80);
медиана DMFS: 41,8 месяца (25,6–55,3) — против 19,4 месяца (11,4–34,4), HR 0,70 (0,55–0,89);
медиана PFS2: 55,4 месяца (40,1–NE) — против 30,0 месяца (24,8–47,1), HR 0,70 (0,55–0,89).
«Китруда» (Keytruda, пембролизумаб), блокатор PD-1 авторства «Мерк и Ко» (Merck & Co.), разрешен для первоочередного лечения рака шейки матки на стадии III–IVA. Назначение пембролизумаба следует осуществлять совместно с химиотерапией и облучением.
Соответствующее решение вынесено Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) в середине января 2024 года.
До этого, в середине октября 2021 года, «Китруда» был одобрен FDA для первоочередного лечения персистирующего, рецидивирующего или метастатического рака шейки матки, опухоли которого экспрессируют PD-L1 (CPS ≥ 1). «Китруда» применяется совместно с химиопрепаратами, а также «Авастином» (Avastin, бевацизумаб) или без него.
Тогда же американский регулятор выдал полноценное разрешение на моноприменение «Китруды» в терапии второй линии. В июне 2018 года «Китруда» был условно дозволен для второлинейного лечения рецидивирующего или метастатического рака шейки матки, который прогрессировал во время или после химиотерапии и опухоли которого характеризуются наличием экспрессии PD-L1 (CPS ≥ 1).
Клиническое исследование KEYNOTE-826 (NCT03635567) фазы III (рандомизированное, двойное слепое, плацебо-контролируемое, многоцентровое, международное) охватило взрослых пациентов (n=617) с персистирующей (стойкой), рецидивирующей или метастатической плоскоклеточной карциномой, аденокарциномой или аденосквамозной карциномой шейки матки, которые прежде не проходили системную химиотерапию (за исключением одновременного применения в качестве радиосенсибилизирующего агента) и более не подходят для радикального лечения, такого как хирургия и/или облучение.
Среди основных характеристик испытуемых: у 89% положительная экспрессия PD-L1 (CPS ≥ 1), при этом у 51% CPS 10 и выше, у 37% CPS 1–10 и у 11% CPS < 1; 63% получали бевацизумаб; медиана возраста 51 год (22–82); плоскоклеточная карцинома у 75%, аденокарцинома у 21%, аденосквамозная карцинома у 5%; заболевание на стадии IVB у 31%, метастатическое заболевание у 20%.
Все участники получали стандартную платиносодержащую химиотерапию (паклитаксел с цисплатином или паклитаксел с карбоплатином) вместе с ингибитором ангиогенеза «Авастином» (Avastin, бевацизумабом) или без него. Экспериментальной группе дополнительно назначали «Китруду».
Подключение блокатора PD-1 привело к статистически значимому продлению выживаемости без прогрессирования (PFS) и общей выживаемости (OS). Указанное оказалось справедливым равно как вне зависимости от статуса опухолевой экспрессии PD-L1, так и без оглядки на использование бевацизумаба.
Промежуточные результаты
Данные, собранные после наблюдений на протяжении медианных 22,0 месяца (15,1–29,4), получились следующими.
В подгруппе пациентов с положительной опухолевой экспрессией PD-L1 (CPS ≥ 1) медиана PFS составила 10,4 месяца (95% ДИ: 9,7–12,3) в группе «Китруды» — против 8,2 месяца (95% ДИ: 6,3–8,5) в группе плацебо (отношение риска [hazard ratio, HR] 0,62 [95% ДИ: 0,50–0,77]; p<0,001).
Медиана OS в группе «Китруды» достигнута не была (95% ДИ: 19,8–NR), тогда как в контрольной группе она вышла к 16,3 месяца (95% ДИ: 14,5–19,4) [HR 0,64 (95% ДИ: 0,50–0,81); p<0,001].
Частота общего ответа (ORR) получилась равной 68% (95% ДИ: 62–74), включая 23% полных ответа (CR) и 45% частичных ответов (PR), — против ORR 50% (95% ДИ: 44–56), в том числе CR 13% и PR 37%.
Медиана длительности ответа (DoR) остановилась на 18,0 месяца (1,3+ — 24,2+) — против 10,4 месяца (1,5+ — 22,0+).
В подгруппе пациентов с усиленной экспрессией PD-L1 (CPS ≥ 10) медиана PFS составила 10,4 месяца (95% ДИ: 8,9–15,1) — против 8,1 месяца (95% ДИ: 6,2–8,8) [HR 0,58 (95% ДИ: 0,44–0,77); p<0,001]. Медиана OS достигнута не была (95% ДИ: 19,1–NR) — против 16,4 месяца (95% ДИ: 14,0–25,0) [HR 0,61 (95% ДИ: 0,44–0,84); p=0,001].
Во всей популяции испытуемых медиана PFS составила 10,4 месяца (95% ДИ: 9,1–12,1) — против 8,2 месяца (95% ДИ: 6,4–8,4) [HR 0,65 (95% ДИ: 0,53–0,79); p<0,001]. Медиана OS вышла к 24,4 месяца (95% ДИ: 19,2–NR) — против 16,5 месяца (95% ДИ: 14,5–19,4) [HR 0,67 (95% ДИ: 0,54–0,84); p<0,001].
Итоговые результаты
По прошествии медианных 39,1 месяца наблюдений итоговое заключение следующее.
Добавление пембролизумаба к химиотерапии с бевацизумабом или без него снижает риск смерти на 40% в популяции пациентов с опухолевой экспрессией PD-L1 на уровне CPS ≥ 1, на 42% — CPS ≥ 10 и на 37% — во всей популяции испытуемых вне зависимости от уровня PD-L1.
Продолжающееся клиническое исследование KEYNOTE-A18 (NCT04221945) фазы III (рандомизированное, двойное слепое, плацебо-контролируемое, многоцентровое, международное) пригласило взрослых пациентов (n=1060) с ранее нелеченным высокорисковым местнораспространенным раком шейки матки (плоскоклеточная карцинома, аденокарцинома или аденосквамозная карцинома).
Под высоким риском подразумевалось заболевание на стадии IB2–IIB с поражением лимфатических узлов или на стадии III–IVA.
Испытуемые были стратифицированы по стадированию рака шейки матки:
на стадии IB2–IIB с поражением лимфатических узлов (n=462): опухолевые поражения больше 4 см или клинически видимые поражения, распространившиеся за пределы матки, но не распространившиеся на стенки таза или нижнюю треть влагалища;
на стадии III–IVA с поражением лимфатических узлов или без такового (n=596): опухолевые поражения нижней части влагалища с распространением или без распространения на боковые стенки таза, гидронефрозом и/или нефункционирующей почкой либо с распространением на соседние органы таза;
на стадии IVB (n=2).
Пациентам назначали либо пембролизумаб на фоне химиотерапии (цисплатин) и облучения (дистанционная лучевая терапия, затем брахитерапия), либо только химиотерапию.
Лечение проводилось до момента прогрессирования заболевания или неприемлемой токсичности.
После наблюдений на протяжении медианных 17,9 месяца (0,9–31,0) добавление «Китруды» к стандартной химиолучевой терапии снизило риск прогрессирования заболевания или смерти на 30% относительно группы сравнения: отношение риска (hazard ratio, HR) 0,70 (95% ДИ [здесь и далее]: 0,55–0,89; p=0,0020).
Медиана выживаемости без прогрессирования (PFS) достигнута не была.
24-месячная частота PFS составила 67,8% в группе пембролизумаба — против 57,3% в контрольной группе.
Показатель общей выживаемости (OS) пока не созрел.
Последующий эксплоративный анализ установил, что наибольший вклад в улучшение PFS внесли пациенты с раком шейки матки на стадии III–IVA: снижение риска прогрессирования заболевания или смерти составило относительных 41% (HR 0,59 [0,43–0,82]).
12-месячная частота PFS вышла к 81% (75–85) — против 70% (64–76).
Среди пациентов с заболеванием на стадии IB2–IIB данный риск уменьшился лишь на относительных 9% (HR 0,91 [0,63–1,31]).
Экспертные комментарии
Рак шейки матки является четверым самым распространенным онкологическим заболеванием в мире и четвертой причиной смерти женщин от опухолевой патологии [1]. В США на метастатической стадии рак шейки матки диагностируется у 16% пациентов [2], и пятилетняя выживаемость при этом составляет 17% [3].
Стандартным первоочередным лечением при персистирующем, рецидивирующем или метастатическом раке шейки матки является платиносодержащая химиотерапия, причем, ввиду баланса между эффективностью и безопасностью, предпочтение отдается схеме, состоящей из препарата платины (цисплатин или карбоплатин), паклитаксела и бевацизумаба [4] [5] [6] [7] [8] [9] [10].
Итоговые результаты клинического испытания KEYNOTE-826 (NCT03635567) надежно доказали оправданность добавления блокатора PD-1 к стандартной схеме лечения персистирующего, рецидивирующего или метастатического рака шейки матки.
Во-первых, подключение иммунотерапии на ранних этапах лечения продемонстрировало существенное продление жизни — в сравнении с переходом к терапии второй линии.
Во-вторых, даже если пациенты проходят лечение без бевацизумаба, иммунотерапия всё равно приносит им пользу. Для справки: ввиду противопоказаний воспользоваться бевацизумабом может лишь ограниченное число больных — не более 15% [11].
В-третьих, иммунотерапия успешно работает вне зависимости от наличия или отсутствия опухолевой экспрессии PD-L1.
«Мерк и Ко» на достигнутом не остановилась. Результаты продолжающегося клинического испытания KEYNOTE-A18 (NCT04221945) фазы III установили, что оправдано добавление «Китруды» к химиолучевой терапии при ранее нелеченном высокорисковом местнораспространенном раке шейки матки. Однако подключение пембролизумаба проявило себя должным образом только среди пациентов с более запущенным заболеванием: на стадии III–IVA, но не на более ранней стадии IB2–IIB.
Такое заболевание характеризуется плохим прогнозом: через два года большинство пациентов сталкивается с рецидивом. Стандартном лечения местнораспространенного рака шейки матки является лучевая терапия с одновременной химиотерапией и последующей брахитерапией. Считается, что иммуностимулирующая активность пембролизумаба должна усилиться на фоне химиолучевой терапии.