ЧТО ПРОИЗОШЛО
Экспериментальный препарат бепировирсен (bepirovirsen), разрабатываемый «ГлаксоСмитКляйн» (GlaxoSmithKline), располагает необходимым потенциалом, для того чтобы обеспечить функциональное излечение хронического вирусного гепатита B.
ОСНОВНЫЕ ФАКТЫ
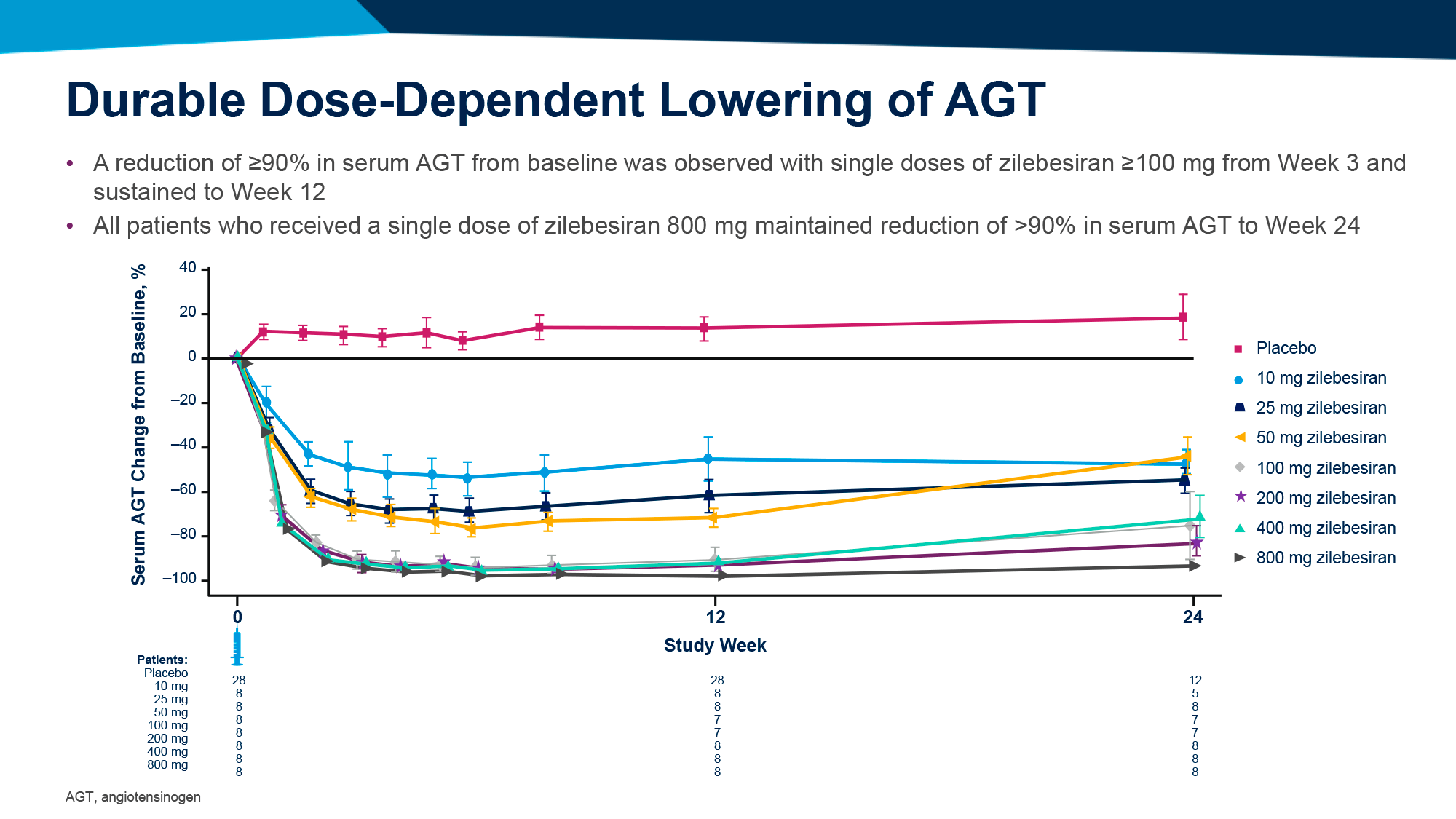
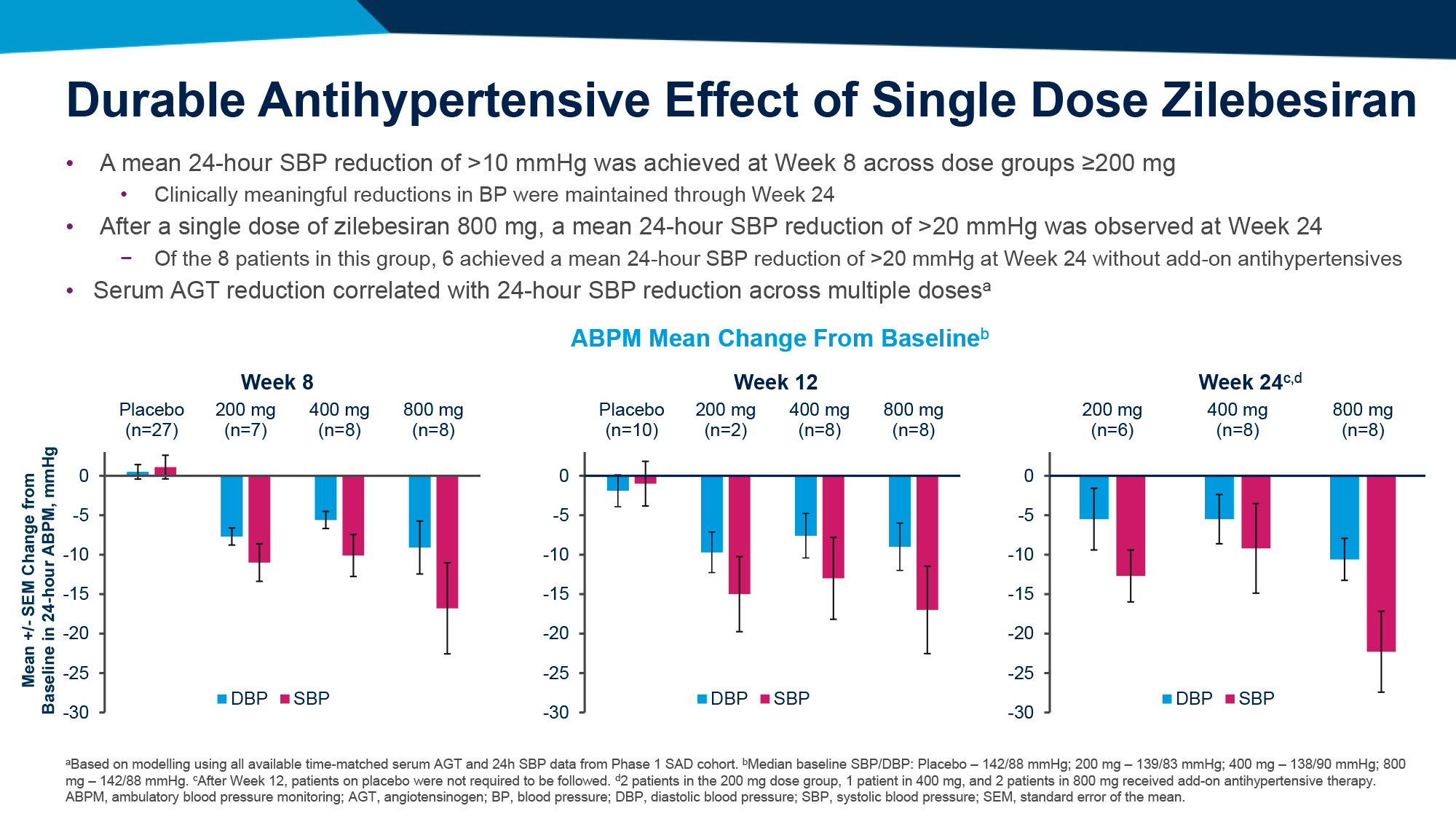
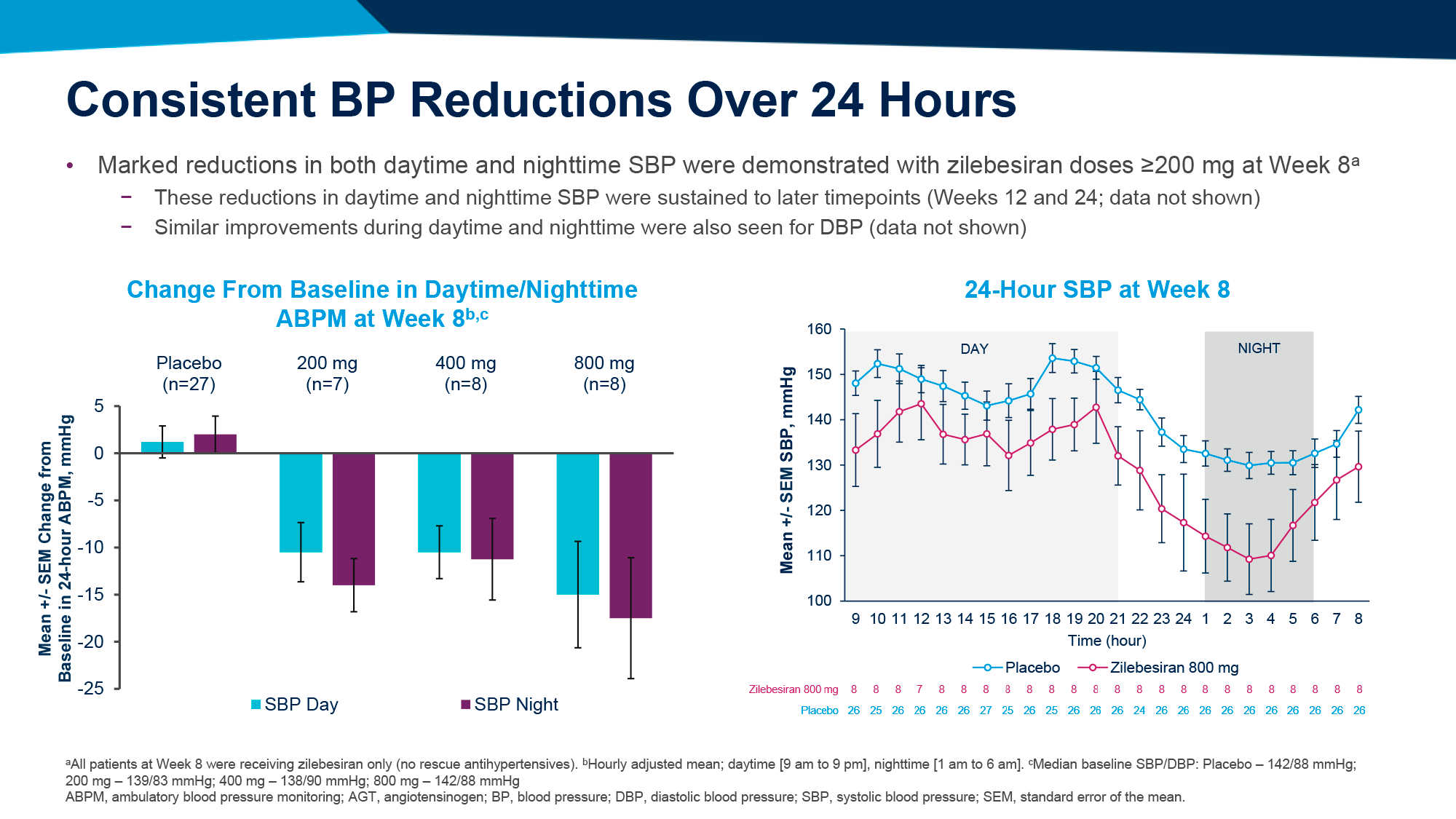
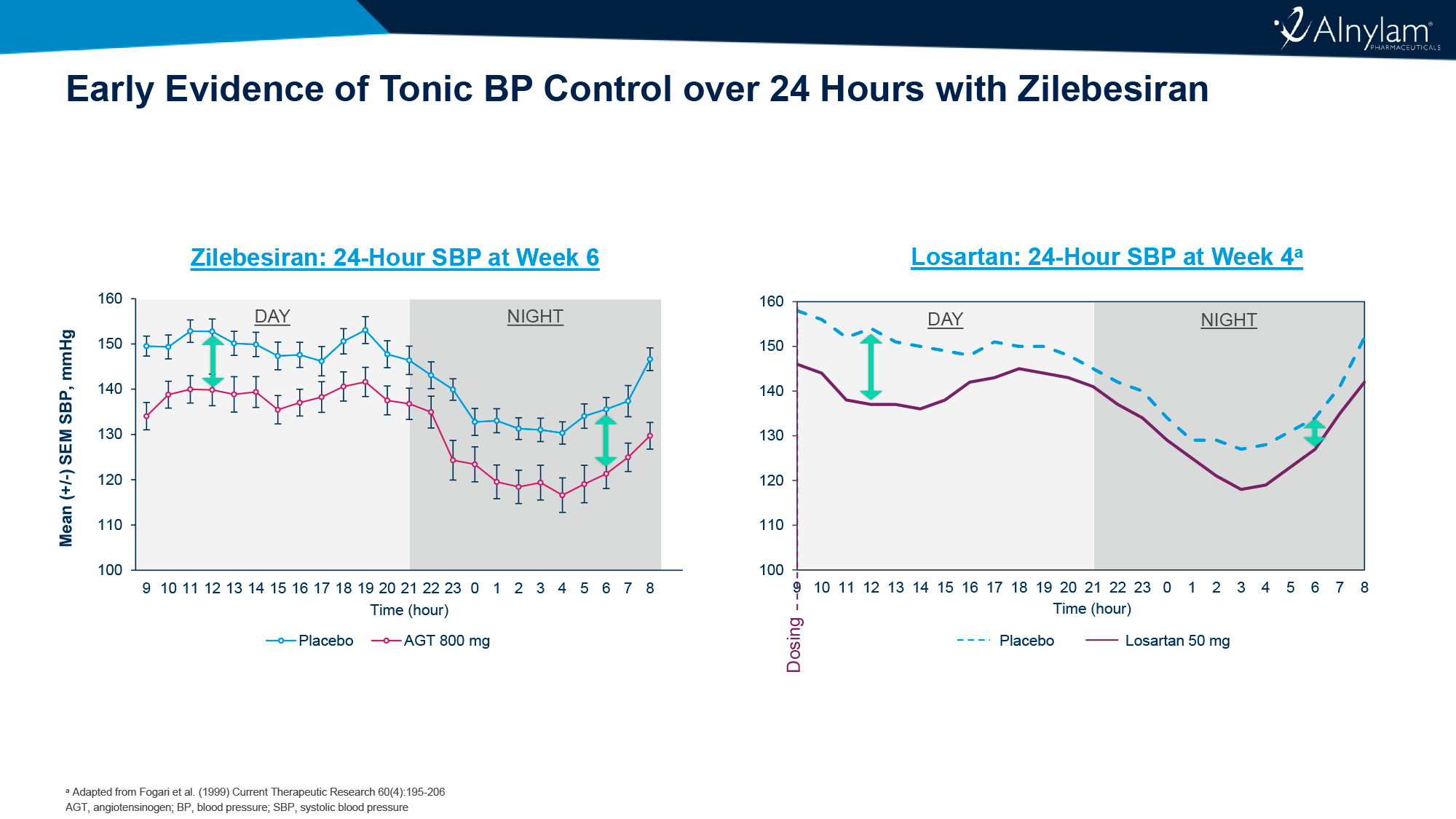
Продемонстрировано, что еженедельные подкожные инъекции бепировирсена привели к полному клиренсу ДНК и поверхностного антигена вируса гепатита В (HBsAg) у почти трети пациентов после 6 месяцев лечения.
Впрочем, по прошествии 6 месяцев наблюдений после завершения терапии статус функционального излечения оказался справедливым для существенно меньшей пропорции больных. Однако терапию всё равно можно назвать успешной, если сравнивать с существующими лекарственными средствами.
В настоящее время хронический вирусный гепатит B, которым заражены 254 млн человек во всём мире, неизлечим: приходится придерживаться пожизненной терапии, которая далеко не всегда оказывается успешной [1].
Напротив, хронический вирусный гепатит C считается полностью излечимым заболеванием. С мая 2011 года стали планомерно появляться всё более эффективные противовирусные препараты прямого действия (ПППД), за несколько месяцев исцеляющие эту инфекционную болезнь.

Лекарства по цене золота: дорогостоящие препараты не способствуют инновациям
Цена лекарств постоянно растет, несмотря на отсутствие значимых улучшений. Разбираемся, почему фармацевтическая индустрия превратилась в машину для выкачивания денег.
КАК ЭТО РАБОТАЕТ
Бепировирсен (bepirovirsen, GSK3228836, IONIS-HBVRx) представляет собой антисмысловой олигонуклеотид (ASO) — одноцепочечную ДНК, которая комплементарна 20 консервативным нуклеотидным последовательностям всех матричных РНК (мРНК) вируса гепатита B (HBV), включая его прегеномную РНК (пгРНК).
Связывание бепировирсена с мРНК и пгРНК HBV приводит к образованию гибридного комплекса, который рекрутирует эндогенную рибонуклеазу H (РНКаза H). Этот фермент расщепляет мРНК и пгРНК HBV, тем самым препятствуя трансляции белков вируса. В результате уменьшается количество РНК, ДНК и белков HBV, в том числе поверхностного антигена вируса гепатита В (HBsAg). Вирусная нагрузка снижается, сдерживаются процессы инфицирования и репликации HBV, появляется шанс на функциональное излечение хронического вирусного гепатита B [1] [2].
Бепировирсен также проявляет агонистическую активность в отношении толл-подобного рецептора 8 (TLR8), тем самым работая как иммуностимулятор, индуцирующий активность врожденного иммунитета и выработку цитокинов [3] [4] [5] [6] [7].
Поскольку бепировирсен не конъюгирован с аминосахаром N-ацетилгалактозамином (GalNAc), нужным для более таргетной доставки в печень, он в большей степени распределяется в непаренхимных клетках печени, нежели в гепатоцитах, и потому распознается резидентными иммунными клетками печени, что приводит к активации сигнализации врожденной иммунной системы [8].
Бепировирсен разработан «Айонис фармасьютикалс» (Ionis Pharmaceuticals), которая в конце августа 2019 года лицензировала его «ГлаксоСмитКляйн» (GlaxoSmithKline). Взамен обещано до 262 млн долларов и роялти от реализации готового лекарственного препарата [9].
Состоятельность терапевтической парадигмы антисмысловых олигонуклеотидов подтверждена немалым числом уже одобренных лекарственных препаратов, направленных на сдерживание экспрессии специфических белков, патогенных в случае какого-либо заболевания. Так, например, «Эксондис 51» (Exondys 51, этеплирсен), «Виондис 53» (Vyondys 53, голодирсен), «Амондис 45» (Amondys 45, касимерсен) и «Вилтепсо» (Viltepso, вилтоларсен) применяются в лечении мышечной дистрофии Дюшенна, «Спинраза» (Spinraza, нусинерсен) используется в лечении спинальной мышечной атрофии, «Тегседи» (Tegsedi, инотерсен) назначается для лечения полинейропатии при наследственном транстиретиновом амилоидозе.
КЛИНИЧЕСКИЕ ПОДРОБНОСТИ
В целях предварительного выяснения эффективности и безопасности лечения хронического вирусного гепатита B при помощи бепировирсена «ГлаксоСмитКляйн» положилась на клиническую программу из трех испытаний фазы II:
- B-Clear (NCT04449029): 24- или 12-недельное назначение бепировирсена (разными схемами), причем либо на фоне терапии нуклеозидными/нуклеотидными аналогами (NA), либо без нее.
- B-Together (NCT04676724): 24- или 12-недельное применение бепировирсена на фоне NA-терапии, за которым следует 24-недельный курс пегилированного интерферона альфа-2a.
Первичная конечная точка эффективности лечения хронического вирусного гепатита B в первых двух клинических испытаниях была установлена пропорцией пациентов, продемонстрировавших устойчивую вирусную супрессию (подавление вирусной нагрузки; SVR), под которой понимают снижение уровней поверхностного антигена вируса гепатита В (HBsAg) и ДНК HBV ниже определяемых высокоточным методом ПЦР порогов (соответственно 0,05 МЕ/мл и 20 МЕ/мл), сохраняющееся на протяжении 24 недель после завершения лечения и при условии отсутствия в этот период дополнительной терапии какими-либо противовирусными препаратами (если таковые ранее не принимались).
- B-Sure (NCT04954859): среди ранее прошедших лечение бепировирсеном и ответивших на него пациентов из других клинических испытаний. В ходе 33-месячных наблюдений выясняется, как долго сохраняется SVR-статус.
B-CLEAR
Клиническое исследование B-Clear (NCT04449029) фазы IIb (рандомизированное, двойное слепое, плацебо-контролируемое, многоцентровое, международное) пригласило взрослых пациентов (n=457) с хроническим вирусным гепатитом B, проходящих либо нет фоновое лечение нуклеозидными/нуклеотидными аналогами (NA).
На протяжении 24 недель участникам еженедельно подкожными инъекциями назначали бепировирсен (разными схемами):
- группа 1: бепировирсен 300 мг — 24 недели;
- группа 2: бепировирсен 300 мг — 12 недель, затем бепировирсен 150 мг — 12 недель;
- группа 3: бепировирсен 300 мг — 12 недель, затем плацебо — 12 недель;
- группа 4: плацебо — 12 недель, затем бепировирсен 300 мг — 12 недель.
Группы 1, 2 и 3 также получили нагрузочные дозы бепировирсена: по 300 мг на 4-й и 11-й дни.
Согласно промежуточному анализу собранных данных, после 24-недельной терапии хронического вирусного гепатита B наилучшую результативность показала группа 1. Так, одновременное отсутствие HBsAg и ДНК HBV зафиксировано для 28% и 29% испытуемых, соответственно придерживавшихся фоновой терапии NA и не принимавших такие препараты. При этом у 68% и 65% участников уровень HBsAg упал ниже 100 МЕ/мл [1].



Итоговые результаты клинического исследования получились следующими [2].
Среди тех пациентов, которые придерживались фоновой терапии NA, к первичной конечной точке эффективности лечения вышли 9%, 9%, 3% и 0% участников в группах 1, 2, 3 и 4. Среди тех, кто не получал NA, первичная конечная точка достигнута среди 10%, 6%, 1% и 0% испытуемых.

Если допустить «всплески» активности вируса гепатита B (однократное повышение HBsAg или ДНК HBV до уровней, превышающих или равных пороговым), то есть несколько ослабить критерии выхода к первичной конечной точке, ее достигли 10%, 9%, 4% и 2% пациентов на фоновой терапии NA и 14%, 6%, 1% и 4% пациентов без фоновой терапии NA.
Успешный ответ на назначение бепировирсена напрямую зависел от исходного уровня HBsAg до начала лечения: он чаще фиксировался у испытуемых с низким уровнем HBsAg (≤ 3 log10 МЕ/мл) и реже с высоким (> 3 log10 МЕ/мл). К примеру, в группе 1 первичная конечная точка была засвидетельствована у 16% и 25% пациентов с низким изначальным уровнем HBsAg и у 6% и 7% пациентов с высоким — соответственно среди получавших и не получавших фоновую терапию NA.
Бепировирсен характеризовался приемлемой переносимостью. Наиболее распространенным нежелательным явлением были реакции по месту введения препарата.
B-TOGETHER
Клиническое исследование B-Together (NCT04676724) фазы IIb (рандомизированное, открытое, многоцентровое, международное) пригласило взрослых пациентов (n=108) с хроническим вирусным гепатитом B.
Испытуемым, продолжающим следовать стабильной терапии нуклеозидными/нуклеотидными аналогами (NA), назначали бепировирсен (еженедельными подкожными 300-мг инъекциями) на протяжении 24 недель (группа 1) или 12 недель (группа 2); плюс нагрузочные дозы бепировирсена (по 300 мг на 4-й и 11-й дни). Далее участники проходили 24-недельный курс пегилированного интерферона альфа-2a (в еженедельной подкожной дозе 180 мкг).
Экспериментальное лечение обеспечило выход к первичной конечной точке для 9% и 15% пациентов в группах 1 и 2. При этом исходный уровень HBsAg коррелировал с пропорцией ответивших на лечение больных. Так, при низком изначальном уровне HBsAg (≤ 1000 МЕ/мл) первичная конечная точка была зафиксирована для 24% и 41% испытуемых, тогда как при высоком его уровне (≤ 3000 МЕ/мл) — для 14% и 26% [1].
Большинство пациентов (58% в каждой группе), показавших ответ на лечение по завершении применения бепировирсена, не столкнулись с рецидивом инфекции в ходе назначения интерфероновой терапии. Другими словами, добавление интерферона снизило риск рецидива хронического вирусного гепатита B.
Однако только 2 человека (в группе 2) с частичным ответом после бепировирсена вышли к полному ответу в процессе добавления интерферона. То есть подключение последнего к бепировирсену не привело к значимому улучшению исходов, обусловленных снижением уровня HBsAg.
Был отмечен временный рост уровня АЛТ выше утроенной верхней границы нормы. В ходе интерфероновой терапии не было отмечено ассоциации между ростом HBsAg и АЛТ.
Серьезные нежелательные явления (НЯ), связанные с лечением, были зарегистрированы только у 1 пациента (2%). Некоторые НЯ вынудили выйти из исследования 4% испытуемых (n=4): аллергический дерматит, реакции по месту введения препарата, депрессия.




B-SURE
Продолжающееся клиническое испытание B-Sure (NCT04954859) фазы II (нерандомизированное, открытое, многоцентровое, международное) поставило своей целью выяснить, как долго сохраняется SVR-статус среди ранее прошедших лечение бепировирсеном и ответивших на него пациентов с хроническим вирусным гепатитом B из других клинических испытаний этого экспериментального препарата. Наблюдательное исследование продолжается сроком максимум 33 месяца.
Положительный SVR-статус разнесен по критерию ответа на бепировирсен: полный ответ (CR) и частичный ответ (PR). Первый, отражающий функциональное излечение хронического вирусного гепатита B, предполагает снижение уровней поверхностного антигена вируса гепатита В (HBsAg) и ДНК вируса гепатита B (HBV) ниже порогов, определяемых высокоточным ПЦР-методом: соответственно 0,05 МЕ/мл и 20 МЕ/мл. Второй — уровень HBsAg < 100 МЕ/мл и уровень ДНК HBV ниже вышеуказанного порогового.
Если говорить о пациентах из B-Clear (NCT04449029), то анализ исходов B-Sure осуществляется согласно разбивке по факту терапии нуклеозидными/нуклеотидными аналогами (NA): одна группа пациентов из B-Clear получала фоновое NA-лечение, но затем, по прошествии 3 месяцев после начала участия в B-Sure, должна была прекратить, тогда как вторая группа вообще не проходила фоновую NA-терапию.
В группе фонового NA-лечения оказались 11 человек с полным ответом, из которых 9 пациентов впоследствии, согласно протоколу исследования, прекратили прием NA-препаратов. По прошествии 6 месяцев после остановки NA-терапии полный ответ сохранился у 78% (n=7/9) участников. По прошествии еще 6 месяцев (всего 12), когда 1 больной был исключен из анализа ввиду недоступности данных наблюдений за ним, полный ответ сохранился у всех оставшихся 6 человек, то есть составил 67% (n=6/9).
Ситуация с пациентами, показавшими частичный ответ к моменту включения в B-Sure, следующая. Из 29 пациентов прием NA-препаратов должным образом прекратили 23 человека. По истечении 6 месяцев частичный ответ сохранился у 22% испытуемых (n=5/23), притом что 13% (n=3/23) продемонстрировали отложенный полный ответ. После 12 месяцев частичный ответ сохранился у 13% (n=3/23), а отложенный полный ответ — у 13% (n=3/23).

В группу, не проходившую фоновое NA-лечение, попали 16 пациентов: 11 человек с полным ответом и 5 с частичным. По прошествии 15 месяцев полный ответ сохранился у 36% (n=4/11), частичный — у 20% (n=1/5).

Таким образом, лечение хронического вирусного гепатита B при помощи бепировирсена способно привести к функциональному излечению этой инфекции, что подтверждается результатами долгосрочных наблюдений за пациентами, прекратившими всякое лечение заболевания.
ЧТО ДАЛЬШЕ
На волне обнадеживающих результатов, продемонстрированных бепировирсеном в задаче излечения хронического вирусного гепатита B, «ГлаксоСмитКляйн» запустила два идентичных опорных клинических испытания, B-Well 1 (NCT05630807) и B-Well 2 (NCT05630820), фазы III (рандомизированных, двойных слепых, плацебо-контролируемых, многоцентровых, международных), которые, если завершатся успешно, лягут в основу регистрационного досье.
Среди основных требований к взрослым участникам (n=900 и n=900): стабильная терапия нуклеозидными/нуклеотидными аналогами (NA) на протяжении не менее чем 6 месяцев; уровень HBsAg в пределах 100–3000 МЕ/мл; уровень ДНК HBV < 90 МЕ/мл; уровень АЛТ не выше удвоенной верхней границы нормы.
Испытуемые, продолжающие следовать фоновой NA-терапии, вначале проходят 24-недельный курс терапии бепировирсеном или плацебо, а затем на протяжении 24 или 48 недель получают только NA-препараты.
Первичная конечная точка эффективности лечения установлена функциональным излечением хронического вирусного гепатита B, факт которого подтверждается устойчивой вирусной супрессией (SVR) на протяжении хотя бы 24-недельного периода, оставляемого без какого-либо лечения. Исследования завершатся ближе к концу 2025 года.
В клиническом исследовании B-United (NCT06537414) фазы IIb пациентам (n=280) с хроническим вирусным гепатитом B, придерживающимся стабильной NA-терапии и находящимся в SVR-статусе, вначале назначают комбинацию из даплусирана (daplusiran) и томлигисирана (tomligisiran) — по 50 и 200 мг каждые 4 недели на протяжении 24 недель, а затем проводят 24-недельный курс лечения бепировирсеном. Результаты испытания, которое должно выяснить частоту функционального излечения инфекции, будут готовы к концу 2027 года.
Комбинация из даплусирана и томлигисирана (JNJ-3989, JNJ-73763989, GSK5637608, ARO-HBV) — фиксированная доза малых интерферирующих РНК (миРНК), таргетированных на гепатоциты и индуцирующих процесс эндогенной интерференции для расщепления транскриптов РНК HBV, экспрессируемых как из ковалентно замкнутой кольцевой ДНК (кзкДНК) HBV, так и из ДНК HBV, интегрированной в геном хозяина. Это приводит к снижению уровня всех белков HBV (HBsAg, HBeAg) и его прегеномной РНК (пгРНК) [1].
Самостоятельно сочетание даплусирана и томлигисирана снижает уровень HBsAg, причем независимо от его исходной концентрации, но обеспечить функциональное излечение хронического вирусного гепатита B не в силах. Антисмысловой олигонуклеотид бепировирсен продемонстрировал свою максимальную эффективность в отношении устойчивой потери HBsAg среди пациентов с изначально относительно низким уровнем HBsAg (≤ 3000 МЕ/мл). Отсюда и родилась гипотеза, что, если снизить уровень последнего перед назначением бепировирсена, можно увеличить пропорцию пациентов, которые выйдут к статусу функционального излечения [2].
В клиническом испытании B-Focus (NCT06497504) фазы II изучается лечение пациентов (n=150) с коинфекцией хронического вирусного гепатита B и вируса иммунодефицита человека 1 (ВИЧ-1). Последний должен находиться в статусе вирусной супрессии благодаря антиретровирусной терапии (АРТ). Результаты будут собраны к середине 2027 года.
ЧТО ЕЩЕ
«ГлаксоСмитКляйн» осуществляет клиническое исследование NCT05276297 фазы II, в котором пациентам (n=184) после 12- или 24-недельной терапии хронического вирусного гепатита B бепировирсеном следует назначение экспериментальной таргетной иммунотерапии GSK3528869A.
Первичная конечная точка эффективности лечения установлена устойчивой вирусной супрессией (SVR) по прошествии 24 недель после терапии. Испытание должно завершиться к зиме 2026 года.
GSK3528869A представляет собой иммунотерапевтическую вакцину из трех компонентов:
- ChAd155-hIi-HBV: лишенный возможности реплицироваться аденовирус шимпанзе группы C серотипа 155, кодирующий последовательности двух белковых антигенов HBV: усеченного ядерного антигена вируса гепатита В (HBcAg) и полноразмерного малого поверхностного антигена вируса гепатита В (S-HBsAg);
- MVA-HBV: кодирующий два вышеуказанных белковых антигена HBV модифицированный осповакцинный вирус Ankara (Modified vaccinia Ankara, MVA), представляющий собой высокоаттенуированный штамм вируса осповакцины (Vaccinia virus);
- HBc-HBs/AS01B-4: вышеуказанные белковые антигены HBV, подкрепленные адъювантом AS01B-4, который представляет собой липосомальное сочетание 3-О-дезацилированного 4′-монофосфорил-липида A (MPL) сальмонеллы (Salmonella minnesota) и молекулы сапонина (QS-21) из растительного экстракта квиллайи мыльной (Quillaja saponaria).
Первый компонент GSK3528869A вводится по завершении курса бепировирсеном: в 1-й день, второй — в 57-й, третий — в 113-й и 169-й.
Концептуальная идея применения терапевтических вакцин вроде GSK3528869A состоит в том, что недостаточность индукции HBV-специфического B- и T-клеточного иммунитета ответственна за отсутствие полного клиренса вируса гепатита B [1] [2] [3] [4]. Вакцина должна запускать формирование сильного вирусоспецифического иммунитета против антигенов HBV, контролирующего инфекцию путем индукции нейтрализующих антител и элиминации инфицированных гепатоцитов при участии эффекторных T-клеток [4] [5].
- В начале декабря 2024 года «ГлаксоСмитКляйн» остановила клиническую проверку NCT03866187 фазы I/II иммунотерапевтической вакцины GSK3528869A, назначаемой пациентам (n=135) с хроническим вирусным гепатитом B, находящимся в SVR-статусе благодаря терапии нуклеозидными/нуклеотидными аналогами (NA). Заявлено об отсутствии должной эффективности по прошествии 24 недель после лечения [6].
Клиническое исследование NCT05330455 фазы I/II, которое должно завершиться к концу 2027 года, тестирует среди пациентов (n=132) с хроническим вирусным гепатитом B сочетание из бепировирсена и GSK3965193, низкомолекулярного ингибитора неканонической атипичной поли(А)-полимеразы 5 и 7 (PAPD5 и PAPD7). Эти ферменты нужны для стабилизации РНК HBV посредством вирусного посттранскрипционного регуляторного элемента (PRE) [7] [8] [9]. Ингибирование PAPD5 и PAPD7 приводит к подавлению вирусной репликации и синтеза вирусных белков, включая HBsAg [10] [11] [12]. В доклинических исследованиях на мышиной модели HBV продемонстрирована оправданность комбинации бепировирсена и GSK3965193 с позиции усиления снижения уровня HBsAg [13].
ЭКСПЕРТНЫЕ КОММЕНТАРИИ
Все существующие стратегии лечения хронического вирусного гепатита B преследуют цель долгосрочной супрессии (подавления) уровня ДНК вируса гепатита B (HBV). При этом весьма желательной является потеря антигена e вируса гепатита B (HBeAg) у HBeAg-положительных пациентов, поскольку она отражает наличие частичного иммунного контроля над инфекцией. В качестве дополнительной цели следует рассматривать нормализацию уровня АЛТ.
Оптимальной конечной точкой лечения выступает устойчивая потеря HBsAg, так как она указывает на глубокую супрессию репликации HBV и экспрессии вирусного белка, свидетельствуя о функциональном излечении (вирусная супрессия на протяжении не менее чем 6 месяцев) хронического вирусного гепатита B, то есть когда вирус не полностью элиминирован (устранен) из организма, но иммунная система контролирует его без каких-либо лекарственных препаратов [1] [2].
Доступные медикаментозные подходы к лечению хронического вирусного гепатита B, представленные пэгинтерфероновой терапией и назначением нуклеозидных/нуклеотидных аналогов (NA), не могут похвастаться безоговорочной эффективностью. Так, потеря HBsAg происходит весьма редко: по прошествии 6 месяцев после годичного курса лечения это наблюдается в 3–7% случаев пэгинтерфероновой терапии и 0–3% случаев терапии NA. Если применение NA продолжается долго, скажем, 5–8 лет, вероятность потери HBsAg повышается, но опять же незначительно: до 10–12% у изначально HBeAg-положительных пациентов и до менее чем 1–2% у HBeAg-отрицательных [1].
Столь скромная эффективность лечения хронического вирусного гепатита B обусловлена тем, что полная эрадикация HBV нынешними препаратами затруднена по причине сохранения в гепатоцитах как ковалентно замкнутой кольцевой ДНК (кзкДНК) HBV, так и интегрированной в их ядро ДНК HBV, являющихся транскрипционными шаблонами для возобновления репликации ДНК HBV [3] [4].
Попытки комбинированного лечения обеспечили потерю HBsAg в 14% случаев, если после минимум 48-недельной NA-терапии переключить пациентов на пэгинтерфероновую терапию [5]. Считается, что прямая противовирусная активность NA, за счет ингибирования вирусной ДНК-полимеразы (обратной транскриптазы) приводящая к вирусологической супрессии и подавлению репликации HBV, частично восстанавливает адаптивный иммунитет, тем самым способствуя улучшению иммуномодулирующего действия пэгинтерферона, проявляющегося в предотвращении образования белков HBV и деплеции (истощении) внутрипеченочного пула кзкДНК [6] [7] [8] [9]. Тем не менее подход нуждается в дополнительных уточняющих исследованиях.
Помимо функционального излечения хронического вирусного гепатита B, существует куда менее достижимая цель стерильного излечения, когда HBsAg не обнаруживается, а ДНК HBV, включая кзкДНК и интегрированную, уничтожена.
В клиническом испытании B-Clear (NCT04449029) бепировирсен продемонстрировал высокую эффективность лечения, если отталкиваться от того факта, что потеря HBsAg установлена для почти трети пациентов после относительно короткого 24-недельного курса лечения.
Вирусологический ответ был зафиксирован среди как HBеAg-отрицательных пациентов, так и придерживающихся NA-терапии HBеAg-положительных. Это свидетельствует о том, что целевая для бепировирсена терапевтическая мРНК-последовательность HBV присутствует даже тогда, когда HBsAg получен из интегрированных вирусных геномов [10].
Отмеченный рост уровня АЛТ, сопутствовавший снижению уровня HBsAg, указывает на благотворные иммунные процессы. Известно, что повышение уровня АЛТ, суррогатного маркера воспаления печени, непрямым образом отражает факт иммуноопосредованного разрушения и клиренса инфицированных гепатоцитов и является предиктором потери HBsAg [11] [12].

Нельзя сказать, что нынешние результаты клинической проверки бепировирсена оказались разочаровывающими. Да, статус функционального излечения хронического вирусного гепатита B, подтвержденный отсутствием HBsAg и ДНК HBV на протяжении 6 месяцев после завершения лечения, зафиксирован у максимум 10% и 14% пациентов — соответственно среди проходивших фоновую терапию NA и без таковой. Однако с учетом короткого курса лечения эффективность следует воспринимать с должным оптимизмом.
Поскольку бепировирсен проявил наибольшую эффективность среди пациентов с изначально низким уровнем HBsAg, в дальнейшем следует рассматривать последний как основополагающий критерий выбора подходящих больных, которые с повышенной вероятностью извлекут пользу от лечения.
Кроме того, одним из предикторов успеха является статус HBeAg. В группе 1 среди HBeAg-отрицательных участников функционально излечились 10% и 14% — соответственно среди проходивших фоновую терапию NA и без таковой. При HBeAg-положительном статусе излечение отмечено для 6% и 0%.
К счастью, наблюдается тенденция, что большинство пациентов с хроническим вирусным гепатитом B являются HBeAg-отрицательными, то есть ДНК-последовательности HBV интегрированы в геном хозяина и являются основным источником HBsAg [10] [13].
Несмотря на множество изучаемых экспериментальных подходов к лечению хронического вирусного гепатита B [14], в их отношении назревает критика: мол, большинство из них фундаментально заблуждаются [15]. Инфекция HBV характеризуется очень высокой степенью генетической пластичности (тысячи квазивидов существуют у каждого отдельного пациента) [16], что является результатом отсутствия коррекционной активности обратной транскриптазы HBV, высокой скорости обновляемости (turnover) кзкДНК и постоянного иммунного давления, оказываемого на вирус. Учитывая, что даже одиночные точечные мутации HBV отменяют способность антисмысловых олигонуклеотидов (ASO) и малых интерферирующих РНК (миРНК) к специфическому расщеплению мРНК [17], под сомнение ставится даже теоретическая состоятельность данных классов лекарственных соединений для лечения хронического вирусного гепатита B.
На вышесказанное намекает тот факт, что снижение уровня HBsAg, обеспеченное бепировирсеном, оказалось сильнее среди пациентов с изначально более низким уровнем HBsAg (≤ 3000 МЕ/мл), что противоречит заявленному механизму действия препарата: сила снижения HBsAg не должна зависеть от его исходного уровня.

В предшествовавшем клиническом испытании NCT02981602 фазы IIa наблюдалась аналогичная картина [18]. Опять же, GSK3389404, вариант бепировирсена, конъюгированный с N-ацетилгалактозамином (GalNAc) в целях более таргетной доставки в гепатоциты [19], не оказал существенного влияния на снижение уровня HBsAg [20].