В середине марта 2024 года Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) одобрило «Ленмелди» (Lenmeldy, атидарсаген аутотемцел) — генную терапию для лечения детей с метахроматической лейкодистрофией с ранним началом: предсимптоматической (без клинических проявлений) поздней инфантильной, предсимптоматической ранней ювенильной или ранней симптоматической ранней ювенильной [1].
Маркетинговое утверждение со стороны Европейского агентства по лекарственным средствам (EMA) состоялось еще в середине декабря 2020 года: препарат получил название «Либмелди» (Libmeldy) [2].
Лечение метахроматической лейкодистрофии при помощи «Ленмелди» / «Либмелди» предполагает однократное вливание генно-терапевтического препарата [3].
«Ленмелди» / «Либмелди» предложен британской «Очед терапьютикс» (Orchard Therapeutics), которую в конце января 2024 года купила японская «Кёва Кирин» (Kyowa Kirin) за 478 млн долларов [4].
ВОПРОС ЦЕНЫ
В США «Очед» сделала «Ленмелди» самым дорогим лекарством современности, зарядив оптовый ценник в размере 4,25 млн долларов, утверждая, что это приемлемая сумма, учитывающая клиническую, экономическую и общественную значимость подобного лечения [1].
Согласно подсчетам Института клинико-экономической экспертизы США (ICER), экономически допустимой является стоимость «Ленмелди» в пределах $2,29–3,94 млн [2].
В Европе «Очед» приходится договариваться с каждой страной отдельно о закупке «Либмелди» за государственный счет на постоянной основе. И потому, во-первых, препарат доступен на очень ограниченном числе территорий [3] и, во-вторых, окончательная цена разнится от страны к стране. Так, к примеру, стоимость «Либмелди» в Германии составила €2,475 млн ($2,690 млн) [4]. Великобритания готова платить £2,875 млн ($3,664 млн), хотя, впрочем, ей предоставлена скидка, размер которой не раскрывается [5]. Ирландия, Бельгия и Нидерланды пришли к соглашению о дисконте в размере 25–65% от исходной суммы в €2,875 млн ($3,129 млн); скидка зависит от подтипа заболевания [6] [7].
ПОЧЕМУ ЭТО ВАЖНО
Метахроматическая лейкодистрофия (MLD), также известная как сульфатидный липидоз, — аутосомно-рецессивная лизосомная болезнь накопления, вызванная биаллельными мутациями гена, кодирующего фермент арилсульфатазу A (ARSA) [1] [2] [3] [4] [5]. Недостаточность ARSA приводит к тому, что в тканях накапливаются сульфатиды, разрушающие миелиновую оболочку нервных волокон, которые перестают функционировать должным образом [6] [7] [8]. Это результирует прогрессирующим психомоторным регрессом, по итогам заканчивающимся полной потерей моторных, поведенческих и когнитивных навыков: пациенты теряют возможность двигаться, говорить, глотать, есть, видеть [9] [10] [11] [12] [13] [14] [15] [16] [17] [18] [19]. Распространенность этого очень редкого и жизнеугрожающего заболевания составляет до 10 случаев на миллион [20] [21] [22] [23] [24].
Метахроматическую лейкодистрофию невозможно полностью вылечить, но аллогенная трансплантация гемопоэтических стволовых клеток (ТГСК) способна приостановить или предотвратить прогрессирование нарушений центральной нервной системы у предсимптоматических или пациентов с ранними симптомами, однако ей сопутствуют риски тяжелых или летальных осложнений [25] [26] [27] [28] [29].
ЧТО ВЫЯСНИЛОСЬ
Эффективность и безопасность генно-терапевтического препарата «Ленмелди» / «Лимбелди» для лечения детей (n=39) с метахроматической лейкодистрофии с ранним началом были изучены в двух клинических испытаниях и программе расширенного доступа (EAP), а затем прошли сравнение с историческими исходами естественного течения болезни у пациентов, оставляемых без лечения. Результаты, сообразно подтипу (форме) заболевания, получились следующими [1].
Предсимптоматическая поздняя инфантильная метахроматическая лейкодистрофия (PSLI MLD) [манифестация заболевания в возрасте до 30 месяцев, отсутствие неврологических признаков и симптомов].
Все дети с PSLI MLD, которые прошли лечение «Ленмелди» / «Лимбелди» и за которыми наблюдали до достижения ими возраста 6 лет и старше, оставались в живых — против 58% в группе сравнения. В возрасте 5 лет 71% испытуемых могли ходить без посторонней помощи и у 85% были нормальные показатели развития языковых и умственных способностей — ничего этого не было в группе сравнения.
Нельзя сказать, что генная терапия «Ленмелди» / «Либмелди» безоговорочно помогла каждому:
Двое детей, которые в возрасте 3,6 и 7,8 лет лишились способности самостоятельно передвигаться, в возрасте 8,1 и 11,6 лет могли передвигаться только с поддержкой.
Один ребенок, столкнувшийся с тяжелыми двигательными нарушениями, к 7,2 годам потерял способности самостоятельно передвигаться и сидеть, а в возрасте 9,9 года полностью утратил двигательную функцию.
Двое детей так и не смогли самостоятельно передвигаться.
Предсимптоматическая ранняя ювенильная метахроматическая лейкодистрофия (PSEJ MLD) [манифестация заболевания в возрасте от 30 месяцев до 7 лет, отсутствие неврологических признаков и симптомов] и ранняя симптоматическая ранняя ювенильная метахроматическая лейкодистрофия (ESEJ MLD) [манифестация заболевания в возрасте от 30 месяцев до 7 лет, самостоятельная ходьба, атаксия, нормальные когнитивные функции (IQ ≥ 85)].
Генно-терапевтическое лечение «Ленмелди» / «Либмелди» детей с PSEJ MLD или ESEJ MLD привело к замедлению процесса ухудшения двигательных навыков, языковых функций и когнитивных способностей. Некоторые дети, столкнувшиеся с прогрессированием заболевания, умерли. В целом необходимы дальнейшие наблюдения, чтобы сделать окончательные выводы.
Среди наиболее распространенных нежелательных явлений в ответ на назначение «Ленмелди» / «Либмелди»: фебрильная нейтропения и стоматит, у большинства проявившиеся в тяжелой форме.
СУТЬ
Согласно систематическому обзору и метаанализу, различия в клинических исходах после лечения поздней инфантильной метахроматической лейкодистрофии (LI MLD) и ранней ювенильной метахроматической лейкодистрофии (EJ MLD) при помощи генной терапии (ГТ) «Ленмелди» / «Либмелди» или аллогенной трансплантации гемопоэтических стволовых клеток (ТГСК), а также при естественном течении (ЕТ) болезни, оставляемой без какого-либо лечения, можно свести к следующему [1].
[table id=66 responsive=»scroll» /]
КАК ЭТО РАБОТАЕТ
Лечение метахроматической лейкодистрофии при помощи генной терапии атидарсаген аутотемцел (atidarsagene autotemcel, OTL-200) обращается к следующим этапам. У пациента осуществляется забор аутологичных гемопоэтических стволовых клеток CD34+, которые ex vivo трансдуцируются лентивирусным вектором, посредством переноса комплементарной ДНК, кодирующей функциональный ген ARSA, и которые затем вливаются обратно в организм. После приживления генетически модифицированных клеток в костном мозге и заселения кроветворного компартмента фермент ARSA, ими продуцируемый и секретируемый, начинает функционировать должным образом, расщепляя сульфатиды и предотвращая их вредное накопление [1] [2].
ЛЮБОПЫТНО
«Ленмелди» — самое дорогое лекарство в мире ($4,25 млн). Ранее в этом звании были такие генно-терапевтические препараты, как «Апстаза» (Upstaza, эладокаген эксупарвовек) [$3,8 млн], «Гемгеникс» (Hemgenix, этранакоген дезапарвовек) [$3,5 млн], «Элевидис» (Elevydis, деландистроген моксепарвовек) [$3,2 млн], «Скайсона» (Skysona, эливалдоген аутотемцел) [$3 млн] и «Роктавиан» (Roctavian, валоктокоген роксапарвовек) [$2,9 млн], предназначенные для лечения недостаточности декарбоксилазы ароматических L-аминокислот, гемофилии B, мышечной дистрофии Дюшенна, церебральной адренолейкодистрофии и гемофилии A.
Следует ли надеяться на появление лекарства за 5 миллионов долларов?
ЧТО ПРОИЗОШЛО. «Трайвио» (Tryvio, апроцитентан) — новый лекарственный препарат, предназначенный для лечения гипертонии у взрослых.
«Трайвио», назначаемый в сочетании с другими гипотензивными лекарственными средствами, применяется, если не удается адекватно контролировать артериальное давление стандартными препаратами.
Апроцитентан (aprocitentan) стал первым новым пероральным гипотензивным препаратом за последние четыре десятка лет, который работает совершенно иначе, чем все существующие лекарства против гипертонии.
«Трайвио», разработанный швейцарской «Айдорсиа фармасьютикалс» (Idorsia Pharmaceuticals), одобрен Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) в середине марта 2024 года [1].
Инструкция по медицинскому применению лекарственного препарата «Трайвио» снабжена «чернорамочным» предупреждением об эмбриофетальной токсичности: апроцитентан нельзя назначать беременным [2].
ПОЧЕМУ ЭТО ВАЖНО. Гипертонией, или артериальной гипертензией, страдает приблизительно 1,4 млрд человек во всём мире [1]. Из них у 10% она характеризуется резистентностью к лечению, когда невозможно добиться должного контроля над уровнем артериального давления даже при одновременном применении трех гипотензивных препаратов различных классов [2]. Постоянно повышенное артериальное давление (≥ 140/90 мм рт. ст.) как следствие резистентной гипертонии резко увеличивает риск неблагоприятных сердечно-сосудистых и почечных событий, таких как болезнь периферических артерий, болезнь коронарных артерий, инсульт, сердечная недостаточность, терминальная стадия почечной недостаточности, смерть по любой причине [3] [4] [5] [6].
ЧТО ВЫЯСНИЛОСЬ. Клиническое испытание PRECISION (NCT03541174) фазы III (рандомизированное, двойное слепое, плацебо-контролируемое, многоцентровое, международное) пригласило взрослых пациентов (n=730), у которых систолическое артериальное давление (САД) было не ниже 140 мм рт. ст., несмотря на прием как минимум трех гипотензивных лекарственных препаратов разных классов (включая диуретик), то есть гипертония была резистентной к лечению.
Перед началом исследования участники, которым ежедневно перорально назначали 12,5 мг апроцитентана или плацебо, были стандартизированы путем перевода на фоновую противогипертоническую терапию, комбинирующую блокатор кальциевых каналов (амлодипин), блокатор рецепторов ангиотензина (валсартан) и диуретик (гидрохлоротиазид). Если ранее использовался бета-блокатор, разрешалось продолжить его применение.
После 4 недель лечения в группе апроцитентана было продемонстрировано усредненное снижение САД и диастолического артериального давления (ДАД) на 15,4 и 10,4 мм рт. ст. — против снижения на 11,6 и 6,4 мм рт. ст. в группе плацебо [1].
Среди наиболее распространенных нежелательных явлений в ответ на назначение апроцитентана: отеки или задержка жидкости (у 9% пациентов), анемия (4%).
СУТЬ. Следует понимать, что снижение систолического артериального давления даже на незначительные, на первый взгляд, 5 мм рт. ст. приводит к уменьшению относительного риска серьезных неблагоприятных сердечно-сосудистых событий (MACE) на 10%. Так, риски инсульта, сердечной недостаточности, ишемической болезни сердца (ИБС) и сердечно-сосудистой смерти снижаются на 13%, 13%, 8% и 5% [1].
КАК ЭТО РАБОТАЕТ. Апроцитентан (aprocitentan, ACT-132577, JNJ-2820) — пероральный низкомолекулярный двойной антагонист рецепторов эндотелина, ингибирующий связывание эндотелина-1 (ET-1) с эндотелиновыми рецепторами A и B (ETA и ETB) [1]. Эндотелин-1 опосредует различные пагубные эффекты, такие как сужение сосудов, фиброз, пролиферация клеток и воспаление. В условиях гипертонии он вызывает дисфункцию эндотелия, гипертрофию и ремоделирование сосудов, активацию симпатической системы, повышение синтеза альдостерона [2] [3].
Апроцитентан является активным метаболитом мацитентана (macitentan), используемого в лечении легочной артериальной гипертензии (ЛАГ) [4].
МЕЖДУ СТРОК. До появления апроцитентана ни одно из существующих системных гипотензивных лекарственных средств не было нацелено на сигнальный путь эндотелина [1]. Имеющийся арсенал препаратов для снижения артериального давления направлен на регуляцию уровня соли и воды, антагонизм ренин–ангиотензин–альдостероновой системы (РААС), снижение притока внеклеточного кальция в клетку, симпатолитическую активность, неселективный вазодилатирующий эффект [2] [3].
ДЛЯ СПРАВКИ. На мировом рынке есть немало антагонистов рецепторов эндотелина (ERA). Так, «Летайрис» / «Волибрис» (Letairis / Volibris, амбризентан), «Опсамит» (Opsumit, мацитентан) и «Траклир» / «Стейвир» (Tracleer / Stayveer, бозентан) применяются в лечении легочной артериальной гипертензии (ЛАГ). «Филспари» (Filspari, спарсентан) предназначен для лечения IgA-нефропатии. «Пивлаз» (Pivlaz, клазосентан) используется для профилактики трех состояний: церебрального вазоспазма, связанного с вазоспазмом инфаркта мозга, церебральных ишемических симптомов после аневризматического субарахноидального кровоизлияния.
ТЕМ ВРЕМЕНЕМ. «АстраЗенека» (AstraZeneca) разрабатывает баксдростат (baxdrostat), пероральный низкомолекулярный ингибитор альдостеронсинтазы, ориентированный на лечение резистентной гипертонии.
ЧТО ПРОИЗОШЛО. «Дувизат» (Duvyzat, гивиностат) — новый лекарственный препарат, предназначенный для лечения мышечной дистрофии Дюшенна у пациентов в возрасте 6 лет и старше.
Гивиностат (givinostat), разработанный итальянской «Италфармако» (Italfarmaco), одобрен Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) в конце марта 2024 года [1] [2].
«Италфармако» была настолько уверена, что FDA вынесет положительный вердикт в отношении «Дувизата», что заблаговременно запустила в США бизнес-подразделение в лице «Ай-ти-эф терапьютикс» (ITF Therapeutics), хотя в конце ноября 2023 года американский регулятор уведомил о переносе срока с принятием решения, ранее установленного на конец декабря 2023-го [3] [4].
В начале сентября 2024 года Европейское агентство по лекарственным средствам (EMA) приступило к рассмотрению регистрационного досье гивиностата [5]. Препарат без каких-либо сомнений получит маркетинговую авторизацию ввиду высокой незакрытой медицинской потребности в новых лекарствах против миодистрофии Дюшенна.
«Дувизат», сделанный в рецептуре пероральной суспензии, необходимо принимать два раза в день.
«Дувизат», который может назначаться на фоне любой терапии, обеспечивает замедление прогрессирования миодистрофии Дюшенна благодаря сдерживанию фиброза и запуску мышечной регенерации.
Среди альтернативных брендовых названий гивиностата: «Дувистат» (Duvistat), «Дувикен» (Duviken), «Дускен» (Dusken), «Увиктос» (Uviktos).
ПОЧЕМУ ЭТО ВАЖНО. Мышечная дистрофия Дюшенна по-прежнему отчаянно нуждается в новых способах лечения. Во-первых, базовая терапия кортикостероидами, такими как преднизолон (prednisolone) / преднизон (prednisone), дефлазакорт (deflazacort) и ваморолон (vamorolone), способствует улучшению двигательной функции, силы и функции легких, задерживает развитие кардиомиопатии, снижает риск развития сколиоза, замедляет потерю способности передвигаться, продлевает выживаемость [1] [2]. Однако неспецифичность кортикостероидов отражается рядом нежелательных явлений, включая увеличение веса, замедление роста, гирсутизм, кушингоидную внешность, переломы, акне, катаракту [1] [3] [4] [5].
Во-вторых, подготовленные «Сарепта терапьютикс» (Sarepta Therapeutics) специализированные лекарственные препараты «Эксондис 51» (Exondys 51, этеплирсен), «Виондис 53» (Vyondys 53, голодирсен) и «Амондис 45» (Amondys 45, касимерсен), а также «Вилтепсо» (Viltepso, вилтоларсен) авторства «Эн-эс фарма» (NS Pharma) в составе японской «Ниппон Синяку» (Nippon Shinyaku) пригодны только для лечения тех пациентов, миодистрофия Дюшенна которых поддается коррекции путем пропуска экзона 51, 53 или 45 соответственно. Эти лекарства, совокупно охватывающие приблизительно 30% популяции больных, некоторые критики называют «изящным с научной точки зрения плацебо»: нет надежных доказательств, что они предоставляют клиническую пользу [6] [7].
В-третьих, генная терапия «Элевидис» (Elevidys, деландистроген моксепарвовек) всё той же «Сарепта» стоит очень дорого.
ЧТО ВЫЯСНИЛОСЬ. Опорное клиническое исследование EPIDYS (NCT02851797) фазы III (рандомизированное, двойное слепое, плацебо-контролируемое, многоцентровое, международное) охватило амбулаторных пациентов (n=179) с мышечной дистрофией Дюшенна в возрасте 6 лет и старше, которые придерживались кортикостероидной терапии и которым на протяжении 18 месяцев дополнительно назначали перорально дважды в день плацебо или гивиностат.
В обеих группах результаты прохождения теста с подъемом по четырем ступеням (4SC) ухудшились относительно исходных: необходимое для его выполнения время увеличилось на 3,03 и 1,25 секунды. Однако разница в 1,78 секунды между группами плацебо и гивиностата оказалась статистически значимой (p=0,037). Другими словами, применение гивиностата привело к замедлению скорости прогрессирования ухудшения мышечных функций [1].
Согласно амбулаторной оценке по системе North Star (NSAA), гивиностат обеспечил 40-процентное замедление прогрессирования заболевания (p=0,0209). Согласно магнитно-резонансной спектроскопии (МРС), назначение гивиностата отразилось 30-процентным уменьшением жировой фракции в латеральной широкой мышце бедра (musculus vastus lateralis) [p=0,0354]: жировая инфильтрация этой мышцы является предиктором потери способности передвигаться. Отмечены положительные тенденции в пользу гивиностата в том, что касается сохранения мышечной силы и результатов теста на время подъема с пола (TTR) и теста 6-минутной ходьбы (6MWT).
Среди наиболее распространенных нежелательных явлений в ответ на назначение гивиностата: диарея (у 36% испытуемых) и рвота (29%).
В предшествовавшем клиническом исследовании NCT01761292 фазы I/II среди амбулаторных пациентов (n=19) с миодистрофией Дюшенна в возрасте 7–10 лет добавление гивиностата к кортикостероидной терапии привело к значительным улучшениям гистологических показателей мышечных тканей, таких как доля площади мышечного волокна (MFAF), площадь поперечного сечения (CSA), некроз, гиперконтрактированные (гиалиновые) волокна, жировое замещение, фиброз (тотальный, эндомизиальный, перимизиальный) [2].
Среди прочих благотворных эффектов гивиностата, принимаемого на протяжении свыше 7 лет: отсрочивание на 3,5 года потери способности самостоятельно ходить и замедление скорости ухудшения легочной функции.
КАК ЭТО РАБОТАЕТ. Гивиностат (givinostat, ITF-2357) — пероральный низкомолекулярный ингибитор гистондеацетилаз (HDAC) классов I и II, располагающий противовоспалительной, антиангиогенной и антинеопластической активностью. Ингибирование HDAC приводит к накоплению высокоацетилированных гистонов с последующей индукцией ремоделирования хроматина и изменением характера экспрессии генов. Гивиностат подавляет выработку провоспалительных цитокинов, таких как фактор некроза опухоли (TNF), интерлейкин 1 (IL-1), интерлейкин 6 (IL-6), интерферон гамма (IFN-γ) [1].
Фармакологическая блокада HDAC, которые конститутивно активны в условиях мышечной дистрофии Дюшенна, сдерживает все процессы, определяющие фиброзное замещение мышц (воспаление, некроз, жировая инфильтрация и фиброз), и стимулирует компенсаторную регенерацию мышечных тканей [2] [3] [4].
ЧТО ДАЛЬШЕ. Продолжается клиническое исследование NCT03373968 фазы II/III, оценивающее долгосрочные безопасность и переносимость гивиностата при назначении пациентам с мышечной дистрофией Дюшенна в возрасте 7 лет и старше. Гивиностат также изучается среди более тяжелых пациентов в ходе клинического исследования ULYSSES (NCT05933057) фазы III, охватывающего неамбулаторных пациентов с миодистрофией Дюшенна в возрасте 9–17 лет.
ЧТО ЕЩЕ. Параллельно «Италфармако» изучает применимость гивиностата в лечении мышечной дистрофии Беккера и истинной полицитемии.
ТЕМ ВРЕМЕНЕМ. В конце июня 2023 года «Сарепта терапьютикс» (Sarepta Therapeutics) предложила «Элевидис» (Elevidys, деландистроген моксепарвовек) — генно-терапевтическое лечение мышечной дистрофии Дюшенна, которое применяется однократно и которое способно кардинальным образом изменить течение этого нейромышечного и смертоносного заболевания путем доставки в организм усеченного, но работающего варианта гена дистрофина, мутации которого ответственны за развитие патологии. Стоимость генной терапии «Элевидис» составляет 3,2 млн долларов.
ЧТО ПРОИЗОШЛО. «Тевимбра» (Tevimbra, тислелизумаб) — новый лекарственный препарат, предназначенный для лечения неоперабельной или метастатической плоскоклеточной карциномы пищевода у взрослых после предшествовавшей системной химиотерапии, которая проводилась без блокатора PD-(L)1.
«Тевимбра», разработанный китайской «Бейджин» (BeiGene), одобрен Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) в середине марта 2024 года [1].
Европейское агентство по лекарственным средствам (EMA) выдало «Тевимбра» аналогичное маркетинговое разрешение чуть раньше — в середине сентября 2023 года [2].
ПОЧЕМУ ЭТО ВАЖНО. Рак пищевода является восьмым по распространенности онкологическим заболеванием в мире и шестой причиной смерти от онкопатологий [1]. Плоскоклеточная карцинома пищевода — самый распространенный подтип рака пищевода: на его долю приходится 85% случаев [2] [3]. Согласно прогнозам, в 2040 году число новых случаев рака пищевода составит 957 тыс., что на 60% больше, чем было зарегистрировано в 2020 году, свидетельствуя о необходимости дополнительных методов лечения [1]. На момент постановки диагноза рака пищевода у свыше двух третей пациентов он имеет прогрессирующий или метастатический характер. Ожидаемая пятилетняя выживаемость при раке пищевода с отдаленными метастазами не превышает 6% [4].
ЧТО ВЫЯСНИЛОСЬ. Клиническое исследование RATIONALE 302 (NCT03430843) фазы III (рандомизированное, открытое, с группой активного сравнения, многоцентровое, международное) охватило взрослых пациентов (n=512) с неоперабельной, местнораспространенной или метастатической плоскоклеточной карциномой пищевода, которая прогрессировала в ходе первоочередного лечения или после него.
Назначение тислелизумаба привело к улучшению исходов по сравнению с применением химиотерапии (паклитаксела, доцетаксела или иринотекана) [1]:
Медиана общей выживаемости (OS): 8,6 месяца — против 6,3 месяца. Риск смерти снизился на относительных 30%: отношение риска (hazard ratio, HR) 0,70 (95% ДИ [здесь и далее]: 0,57–0,85; p=0,0001).
Вероятность остаться в живых на протяжении 12 месяцев: 37% — против 24%.
Частота общего ответа (ORR): 20%, включая 2% полных ответов (CR) и 18% частичных ответов (PR), — против 9,8%, включая 0,4% CR и 9,4% PR.
Медиана длительности ответа (DoR): 7,1 месяца — против 4,0 месяца.
У пациентов с показателем PD-L1-позитивности опухолевой площади (TAP) ≥ 10% медиана OS составила 10,3 месяца — против 6,8 месяца: HR 0,54 (0,36–0,79; p=0,0006).
С нежелательными явлениями (НЯ), вызванными лечением и протекавшими в тяжелой форме, столкнулись 19% пациентов в группе тислелизумаба — против 56% в группе химиотерапии.
КАК ЭТО РАБОТАЕТ. Тислелизумаб (tislelizumab, BGB-A317) — гуманизированное моноклональное IgG4-антитело, которое связывает PD-1 на T-клетках, тем самым препятствуя его взаимодействию со своими лигандами, PD-L1 и PD-L2. В итоге разблокируются противоопухолевый иммунный ответ, пролиферация T-клеток и выработка цитокинов. Некоторые опухоли характеризуются повышенной регуляцией лигандов PD-1, что способствует подавлению активного T-клеточного иммунного надзора [1].
Тислелизумаб спроектирован так, чтобы минимально связывать Fc-гамма-рецептор 1 (FcγRI) на макрофагах — в целях ограничения антителозависимого клеточного фагоцитоза (ADCP) T-клеток макрофагами как потенциального механизма резистентности к анти-PD-1-терапии [2] [3] [4].
Тислелизумаб обладает более высоким сродством к PD-1, чем «Китруда» (Keytruda, пембролизумаб) и «Опдиво» (Opdivo, ниволумаб), блокаторы PD-1 авторства «Мерк и Ко» (Merck & Co.) и «Бристол-Майерс Сквибб» (Bristol-Myers Squibb), что может быть частично объяснено различными механизмами связывания с PD-1 [5] [6] [7].
ЧТО ДАЛЬШЕ. «Тевимбра» готовится расширить список показаний, подключив первоочередное лечение неоперабельной, рецидивирующей, местнораспространенной или метастатической плоскоклеточной карциномы пищевода и местнораспространенной неоперабельной или метастатической аденокарциномы желудка или пищеводно-желудочного соустья. К июлю и декабрю 2024 года FDA должно вынести соответствующие вердикты [1] [2].
EMA собирается выдать маркетинговое разрешение «Тизвени» (Tizveni, тислелизумаб) для лечения местнораспространенного или метастатического немелкоклеточного рака легкого (НМРЛ) по трем показаниям: в рамках первочередной терапии неплоскоклеточного и плоскоклеточного НМРЛ в сочетании с химиопрепаратами и в ходе монотерапии второй линии после провала платиносодержащей химиотерапии [3].
ТЕМ ВРЕМЕНЕМ. Дебют тислелизумаба в родном Китае состоялся в конце декабря 2019 года: «Байдзеань» (Baizean) предназначен для лечения рецидивирующей или рефрактерной классической лимфомы Ходжкина после провала как минимум двух линий химиотерапии [1] [2]. Впоследствии тислелизумаб прилично расширил список показаний, выступая своего рода местным аналогом «Китруды», который сейчас охватывает 35 показаний (17 типов опухолей и 2 показания вне привязки к анатомической локализации опухоли) и не собирается на этом останавливаться [3] [4]. На конец 2023 года тислелизумаб был одобрен для применения при 12 показаниях, в 2024 году ожидается добавление еще трех [5].
ОБРАТНАЯ СТОРОНА. Идея фикс тислелизумаба, который целиком и полностью придуман в Китае, состоит в том, чтобы избавить эту коммунистическую страну от кабалы дорогостоящих западных блокаторов PD-(L)1 [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11] [12] [13] [14]. Рано или поздно, но это обязательно случится, тем паче эффективность тислелизумаба не уступает американским лекарствам [15] [16] [17] [18] [19] [20] [21] [22] [23] [24] [25] [26] [27] [28] [29].
Китай буквально в приказном порядке вынуждает иностранных и местных фармпроизводителей резко сбрасывать ценники на лекарства, чтобы они имели хождение и страховое медицинское покрытие на столь лакомом ввиду своей масштабности рынке сбыта. И, что примечательно, на правительством уровне поощряет бурное развитие биотехнологий для разработки оригинальных препаратов, а не дженериков. Нет никак сомнений в серьезности, и главное, оправданности намерений: одних только блокаторов PD-(L)1 появилось 15 наименований за минувшие шесть лет, при этом самый дешевый стоит в 16 раз меньше, чем «Китруда» [30].
Могло бы показаться, что появление тислелизумаба на высокоприбыльных Западных рынках наделит «Бейджин» внушительным заработком. Но это не так, ведь в тех же США уже доступны девять блокаторов PD-(L)1. Китайскому фармпредприятию было бы правильным либо придерживаться стратегии сниженных цен, либо выбирать те показания, для которых препараты этого класса еще не одобрены. К примеру, «Кохерус байосайенсиз» (Coherus BioSciences) и «Шанхай Цзюньши байосайенсиз» (Shanghai Junshi Biosciences) выставили стоимость PD-1-блокатора «Локторзи» (Loqtorzi, торипалимаб) на 20% ниже, чем цена «Китруды» [31], и сориентировали его на лечение карциномы носоглотки, для которой прежде не было предложено иммунотерапии [32].
«Рездифра» (Rezdiffra, ресметиром) — новый лекарственный препарат, предназначенный для лечения неалкогольного стеатогепатита (НАСГ) с умеренно-тяжелым фиброзом печени (на стадии F2–F3).
«Рездифра», ориентированный на лечение взрослых пациентов, предполагает терапию НАСГ на фоне изменения образа жизни, включающего соблюдение диеты и повышенную физическую активность.
«Рездифра» не предназначен для лечения НАСГ с циррозом печени.
«Рездифра», разработанный «Мадригал фармасьютикалс» (Madrigal Pharmaceuticals), одобрен Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) в середине марта 2024 года. Регуляторный вердикт вынесен в условном порядке, то есть препарату предстоит подтвердить собственную эффективность.
В начале марта 2024 года Европейское агентство по лекарственным средствам (EMA) приступило к рассмотрению регистрационного досье ресметирома.
«Рездифра» — первое лекарственное средство против НАСГ, появившееся в США.
Ресметиром (resmetirom), относящийся к тиреомиметикам, представляет собой пероральный низкомолекулярный частичный агонист бета-рецептора трийодтиронина (THR-β).
Ряд позднестадийных клинических испытаний подтвердил терапевтическую состоятельность ресметирома для лечения НАСГ. Во-первых, согласно печеночным биоптатам, зафиксированы статистически значимые гистологические улучшения, с достаточной степенью вероятности предсказывающие клиническую пользу. Во-вторых, положительные результаты неинвазивных анализов подкрепили доверие к собранным данным. В-третьих, ресметиром характеризовался приемлемым профилем безопасности: в начале лечения диарея и тошнота легкой степени выраженности.
Что важно, для начала лечения НАСГ ресметиромом не требуется проведения биопсии печени — инвазивной процедуры, ограничивающей доступ к препарату.
В США «Мадригал» установила стоимость оптовой закупки (WAC) годового курса препарата «Рездифра», который необходимо принимать каждый день, на уровне 47,4 тыс. долларов без учета скидок и дисконтов.
Согласно оценкам Института клинико-экономической экспертизы США (ICER), ресметиром экономически эффективен, если окончательная стоимость годового курса лечения укладывается в диапазон от 39,6 тыс. до 50,1 тыс. долларов.
«Мадригал» уверена в светлом будущем ресметира: только в США насчитывается 1,5 млн человек с диагнозом НАСГ, из них у 525 тыс. выраженный фиброз печени (на стадии F2–F3), из которых 315 тыс. проходят врачебное наблюдение.
Согласно прогнозам отраслевых экспертов, мировые продажи «Рездифра» достигнут 2,6–3,5 млрд долларов в 2030 году, а пиковые могут составить 5,5 млрд долларов.
Щитовидная железа производит два тиреоидных гормона: тироксин (T4) и трийодтиронин (T3). Они оказывают мощные эффекты на клеточное развитие, рост и метаболизм и критически необходимы для нормального функционирования почти всех тканей. Стимулирование выработки гормонов щитовидной железы отражается снижением сывороточного холестерина липопротеинов низкой плотности (ЛПНП), усилением основного метаболизма и потерей массы тела [1].
Этот процесс можно контролировать путем таргетирования рецепторов тиреоидных гормонов (THR). Разработка подобных тиреомиметиков сопряжена с проблемой селективности: лекарственное соединение должно быть высокоизбирательным и затрагивать исключительно бета-рецепторы трийодтиронина (THR-β), поскольку агонизм альфа-рецепторов трийодтиронина (THR-α), экспрессирующих в головном мозге, сердечной, почечной, мышечной и костной тканях, приводит к нежелательным явлениям в виде гипертрофии миокарда и усиления дыхания [2].
THR-β, экспрессируемые в том числе в гепатоцитах, отвечают за регуляцию метаболических путей в печени, нарушенных при неалкогольной жировой болезни печени (НАЖБП) и неалкогольном стеатогепатите (НАСГ). Дисфункция THR-β приводит к ослаблению функций митохондрий и митохондриального бета-окисления жирных кислот в сочетании с усилением фиброза [3].
Исследования на животных показали, что THR-β играют важную роль в снижении уровня триглицеридов и холестерина, улучшении чувствительности к инсулину, стимулировании регенерации печени и снижении апоптоза. Имеются данные о том, что НАСГ может быть частично обусловлен снижением уровня тиреоидных гормонов в печени или печеночным гипотиреозом, а частота клинического и субклинического гипотиреоза выше у пациентов с НАЖБП или НАСГ по сравнению с сопоставимыми контрольными группами [3] [4].
Ресметиром (resmetirom, MGL-3196) — пероральный низкомолекулярный частичный агонист THR-β. Ресметиром характеризуется высокой тропностью к гепатоцитам, то есть отсутствует побочное действие на центральную тиреоидную ось: нет в данном случае неблагоприятного эффекта взаимодействия с THR-α. Ресметиром приблизительно в 28 раз более селективен к THR-β, чем THR-α. Ресметиром располагает высокой степенью связывания с белками (> 99%), плохо проникает в ткани вне печени, демонстрирует специфическое поглощение в печени [5].
На доклинических животных моделях НАСГ было установлено, что аналоги тиреоидных препаратов, включая ресметиром, снижают уровень печеночных триглицеридов, стеатоза, перекисного окисления липидов, биомаркеров воспаления и фиброза, а также аланинаминотрансферазы (АЛТ) [3] [5].
В условиях НАСГ селективный агонизм THR-β способствует опосредованным печенью метаболическим эффектам тиреоидных гормонов, таким как уменьшение избытка печеночного жира и снижение уровней атерогенных липидов и липопротеинов, в том числе холестерина ЛПНП, триглицеридов, аполипопротеина B (apoB), липопротеина (a) [(Lp(a)], аполипопротеина CIII (apoCIII), — и всё это без нежелательных системных эффектов, оказываемых посредством THR-α на сердце и кости чрезмерным количеством тиреоидных гормонов [6].
«Рездифра»: эффективность и безопасность ресметирома
Клиническая программа
«Мадригал фармасьютикалс» (Madrigal Pharmaceuticals) организовала клиническую программу MAESTRO фазы III, призванную всецело подтвердить эффективность и безопасность ежедневно назначаемого перорального ресметирома (resmetirom) для лечения взрослых пациентов с неалкогольным стеатогепатитом (НАСГ) [1].
MAESTRO-NASH (NCT03900429): ресметиром (80 или 100 мг) против плацебо — пациенты (n=955) с НАСГ и фиброзом, подтвержденными биопсией. Долгосрочное исследование продолжается сроком до 54 месяцев. Среди основных критериев участия:
один из факторов, подтверждающих НАСГ:
уровень N-концевого пропептида коллагена 3-го типа (PRO-C3) >14 нг/мл или показатель теста Enhanced Liver Fibrosis (ELF) (количественный анализ циркулирующих компонентов внеклеточного матрикса, таких как гиалуроновая кислота, N-терминальный пропептид проколлагена III типа [PIIINP] и тканевой ингибитор 1 металлопротеиназ [TIMP-1]) ≥9;
на аппарате FibroScan: жесткость печени (LSM) для оценки стадии фиброза и контролируемый параметр затухания (CAP), отражающий степень стеатоза печени: соответственно ≥8,5 кПа и ≥280 дБ/м;
биопсия печения, взятая не позднее 2 лет до рандомизации, демонстрирующая фиброз на стадии 1B, 2 или 3.
содержание жира в печени ≥8%, согласно протонной плотности жировой фракции печени, оцененной магнитно-резонансной томографией (MRI-PDFF);
биопсия печения, взятая не позднее 6 месяцев до рандомизации, свидетельствующая о фиброзе на стадии 1A/1C, 1B, 2, или 3, а также общий балл активности неалкогольной жировой болезни печени (НАЖБП) [NAS] ≥4 со всеми положительными (минимум 1 балл) NAS-компонентами (стеатоз, баллонирующая дегенерация, лобулярное воспаление).
MAESTRO-NAFLD-1 (NCT04197479): ресметиром (80 или 100 мг) против плацебо на протяжении 52 недель— пациенты (n=972) с НАЖБП или предположительно НАСГ, в том числе с компенсированным циррозом.
52-недельное исследование позиционируется проводимым в условиях реальной клинической практики, поскольку диагноз ставился на базе неинвазивных показателей, а не биопсии печени.
Среди основных критериев включения один из факторов, подтверждающих наличие заболевания:
LSM ≥5,5 и <8,5 кПа и CAP ≥280 дБ/м;
магнитно-резонансная эластография (MRE) ≥2 и <4,0, MRI-PDFF ≥8% со стеатозом и фиброзом на стадии F1–F3;
биопсия печения, взятая не позднее 2 лет до рандомизации, свидетельствующая о стеатозе и фиброзе.
MAESTRO-NAFLD-OLE (NCT04951219): ресметиром (80 или 100 мг) против плацебо — продолжающееся открытое 52-недельное исследование пригласило (n=1000) пациентов из MAESTRO-NAFLD-1, пожелавших продлить лечение ресметиромом, а также участников из MAESTRO-NASH, проваливших скрининг (NAS < 4).
MAESTRO-NASH-OUTCOMES (NCT05500222): ресметиром (80 мг) против плацебо — продолжающееся сроком до 3 лет исследование вовлекло пациентов (n=700) с ранним циррозом по причине НАСГ.
Поставлена задача неинвазивного отслеживания прогрессирования НАСГ до развития печеночной декомпенсации у пациентов с хорошо компенсированным циррозом.
Определена композитная конечная точка эффективности лечения, установленная временем до наступления одного из следующих событий: смерть по любой причине, трансплантация печени, асцит, печеночная энцефалопатия, кровотечение при варикозно расширенных венах пищевода и желудка, рост балла по модели для оценки терминальной стадии заболеваний печени (MELD) от < 12 до ≥ 15.
Исследование необходимо для реализации двух целей: полноценного одобрения ресметирома для лечения нецирротического НАСГ и расширения популяции пригодных пациентов за счет добавления лечения НАСГ с компенсированным циррозом.
MAESTRO-NASH
Назначение ресметирома на протяжении 52 недель обеспечило выход к двум первичным конечным точкам эффективности лечения неалкогольного стеатогепатита (НАСГ), заявленным, во-первых, пропорцией пациентов, продемонстрировавших разрешение НАСГ (балл баллонирования 0, бал воспаления 1) со снижением общего балла активности неалкогольной жировой болезни печени (НАЖБП) [NAS] минимум на 2 пункта и без ухудшения стадии фиброза и, во-вторых, пропорцией пациентов, показавших улучшение фиброза хотя бы на одну стадию и без ухудшения NAS.
К первой конечной точке вышли 26% и 30% пациентов, получавших 80 и 100 мг ресметирома (resmetirom), — против 10% в группе плацебо (p<0,0001) [1] [2] [3].
Выход ко второй конечной точке был зарегистрирован для 24% и 26% участников — против 14% (p=0,0002 и p<0,0001).
Среди благотворных изменений прочих показателей, обеспеченных ресметиромом и подтвержденных биопсией печени:
снижение балла NAS на ≥ 2 пункта (со снижением балла баллонирования или воспаления на ≥ 1 пункт) и без ухудшения стадии фиброза: у 41% и 45% пациентов, получавших 80 или 100 мг ресметирома, — против 21% (p<0,0001) в группе плацебо;
снижение балла NAS на ≥ 2 пункта (со снижением балла баллонирования или воспаления на ≥ 1 пункт) и с улучшением стадии фиброза: 19% и 21% — против 9%;
улучшение фиброза на 2 стадии (пациенты с фиброзом на стадии F2–F3 с улучшением фиброза на ≥ 2 стадии) и без ухудшения балла NAS: 8% и 10% — против 3% (p=0,0001 и p<0,0001);
разрешение НАСГ и улучшение фиброза на ≥ 1 стадию: 14% и 16% — против 5% (p<0,0001);
разрешение НАСГ (со снижением балла NAS на ≥ 2 пункта и без ухудшения стадии фиброза) [данные наблюдений; как исходный уровень, так и две биопсии]: 32% и 39% — против 11% (p<0,0001);
улучшение фиброза на ≥ 1 стадию и без ухудшения балла NAS [данные наблюдений; как исходный уровень, так и две биопсии]: 30% и 34% — против 16% (p<0,0001);
разрешение НАСГ или улучшение стадии фиброза: 42% и 50% — против 19%.
Изменения отдельных компонентов NAS:
баллонирование: улучшение у 60% и 66% пациентов в группах 80- и 100-мг ресметирома — против 31% в контрольной группе;
воспаление: 46% и 48% — против 32%;
стеатоз: 61% и 68% — против 31%.
Изменения уровней липидов и липопротеинов:
холестерин липопротеинов низкой плотности (ЛПНП): −14% и −20% в группах 80- и 100-мг ресметирома — против −0% в группе плацебо;
аполипопротеин B (ApoB): −16% и −22% — против +1%;
триглицериды (при исходном уровне > 150 мг/дл): −23% и −28% — против −4%;
липопротеин (a) [Lp(a)] (при исходном уровне > 10 нмоль/л): −35% и −38% — против −5%;
аполипопротеин CIII (ApoC-III): −10% и −17% — против +10%;
холестерин липопротеинов невысокой плотности (ЛПНВП): −16% и −22% — против −0%.
Назначение ресметирома привело к существенному снижению уровней печеночных ферментов: аланинаминотрансферазы (АЛТ), аспартатаминотрансферазы (АСТ), гамма-глутамилтрансферазы (ГГТ).
Ресметиром положительно повлиял на печень и селезенку:
содержание жира в печени, согласно протонной плотности жировой фракции печени, оцененной магнитно-резонансной томографией (MRI-PDFF): −42% и −51% в группах 80- и 100-мг дозы ресметирома — против −10% в группе плацебо;
большинство пациентов (> 70%), получавших 100-мг дозу ресметирома, отметились снижением MRI-PDFF на ≥ 30% (медиана снижения 52%), что было прочно ассоциировано с разрешением НАСГ (у 96% пациентов) и улучшением стадии фиброза (88%).
контролируемый параметр затухания (CAP) на аппарате FibroScan (транзиентная эластография), оценивающий показатель жирового перерождения печени, что отражает степень ее стеатоза: −40% и −41% — против −15%;
жесткость печени (LSM), связанная с рубцеванием ее тканей, на аппарате FibroScan с разбивкой по изначальной стадии фиброза (кПа):
F1B: −3,7 и −3,7 — против −0,6;
F2: −2,4 и −2,5 — против −1,3;
F3: −2,0 и −3,3 — против −1,1.
объем печени: −22% и −26% — против −1%;
объем селезенки: −6% и −6% — против +3%.
Зафиксировано снижение веса на ≥ 5% среди 15% и 18% пациентов — против 11%.
Среди наиболее распространенных нежелательных явлений (НЯ) в ответ на назначение ресметирома: со временем преходящие диарея в легкой форме (у 28% и 34% пациентов — против 16% в группе плацебо) и тошнота легкой степени выраженности (22%, 19%, 13%).
Частоты серьезных НЯ были схожими в группах лечения: 12%, 13%, 12%. Частоты прекращения терапии из-за НЯ были низкими: 2%, 7%, 3% — в основном по причине желудочно-кишечных побочных реакций.
MAESTRO-NAFLD-1
Назначение ресметирома (resmetirom) на протяжении 48 недель привело к следующим изменениям метаболических показателей [1]:
холестерин липопротеинов низкой плотности (ЛПНП): −11% и −13% в группах 80- и 100-мг ресметирома — против −1% в группе плацебо (p=0,0004 и p<0,0001);
холестерин ЛПНП (при исходном уровне ≥ 100 мг/дл): −17% и −22% — против −6% (p<0,0001).
аполипопротеин B (ApoB): −14% и −16% — против −1% (p<0,0001);
ApoB (при исходном уровне ≥ 100 мг/дл): −19% и −23% — против −6% (p<0,0001).
триглицериды (при исходном уровне ≥ 150 мг/дл): −23% и −23% — против +0,3% (p<0,0001);
липопротеин (a) [Lp(a)] [при исходном уровне > 10 нмоль/л]: −24% и −34% — против −4% (p<0,0001);
аполипопротеин CIII (ApoC-III): −10% и −14% — против +8% (p<0,0001).
По прошествии 52 недель изменения печеночных показателей оказались следующими:
аланинаминотрансфераза (АЛТ) [при исходном уровне ≥ 30 МЕ/л]: −11% и −12% в группах 80- и 100-мг ресметирома — против −1% в группе плацебо (p=0,0004 и p<0,0001);
аспартатаминотрансфераза (АСТ): −4% и −3% — против +1% (p=0,0141 и p=0,0341);
гамма-глутамилтрансфераза (ГГТ): −10% и −12% — против −2% (p=0,0157 и p=0,0049);
содержание жира в печени, согласно протонной плотности жировой фракции печени, оцененной магнитно-резонансной томографией (MRI-PDFF): −39% и −44% — против −10% (p<0,0001);
контролируемый параметр затухания (CAP) на аппарате FibroScan (транзиентная эластография), оценивающий показатель жирового перерождения печени, что отражает степень ее стеатоза: −37% и −43% — против −18% (p<0,0001);
жесткость печени (LSM), связанная с рубцеванием ее тканей, на аппарате FibroScan (кПа; при исходном уровне ≥ 7,2 кПа): −1 и −2 — против −1 (p=0,6614 и p=0,1710).
Среди наиболее распространенных нежелательных явлений (НЯ) в ответ на назначение ресметирома: со временем преходящие диарея в легкой форме (у 24% и 31% пациентов — против 14% в группе плацебо) и тошнота легкой степени выраженности (12%, 18%, 8%).
Частоты серьезных НЯ были схожими в группах лечения: 6%, 7%, 6%. Частоты прекращения терапии из-за НЯ были низкими: 2%, 3%, 1% — в основном по причине НЯ со стороны желудочно-кишечного тракта.
«Вегови» (Wegovy, семаглутид) отныне можно применять для снижения риска серьезных нежелательных сердечно-сосудистых событий (сердечно-сосудистой смерти, нелетального инфаркта миокарда, нелетального инсульта) у взрослых пациентов с имеющимся сердечно-сосудистым заболеванием при наличии ожирения или избыточной массы тела.
Расширение списка показаний «Вегови», за которым стоит «Ново Нордиск» (Novo Nordisk), одобрено Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) в начале марта 2024 года.
«Вегови», появившийся в начале июня 2021 года, назначается еженедельными подкожными инъекциями для долгосрочной коррекции веса у пациентов в возрасте 12 лет и старше, страдающих либо ожирением, либо избыточной массой тела с сопутствующим лишнему весу заболеванием.
Семаглутид поможет сбросить 15% лишнего веса за год. И даже больше.
Ранее «Ново Нордиск» доказала, что «Вегови» успешно справляется с лечением сердечной недостаточности с сохраненной фракцией выброса среди пациентов с ожирением.
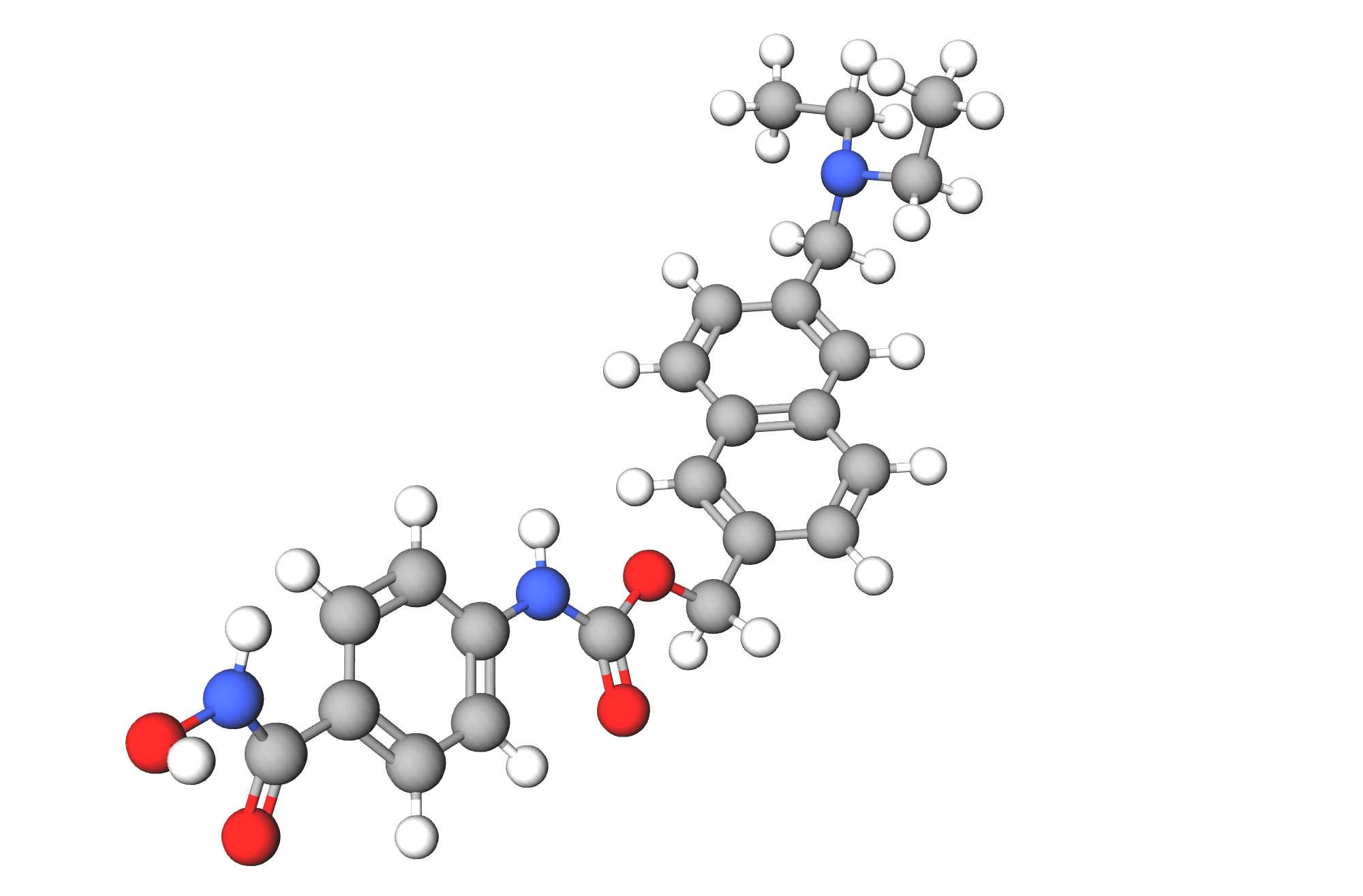
Семаглутид (semaglutide), агонист рецептора глюкагоноподобного пептида-1 (GLP1RA), влияет на широкий спектр метаболических путей, связанных с метаболизмом глюкозы, энергетическим гомеостазом и воспалением.
Дебют семаглутида состоялся в начале декабря 2017 года в лице препарата «Оземпик» (Ozempic, семаглутид), предназначенного для улучшения гликемического контроля при сахарном диабете 2-го типа. Впоследствии «Оземпик» продемонстрировал, что попутно снижает риск серьезных нежелательных сердечно-сосудистых событий при наличии сердечно-сосудистого заболевания и сдерживает прогрессирование существующей хронической почечной недостаточности.
Novo Nordisk предложила инъекционный семаглутид — агонист GLP-1, который эффективнее, чем «Трулисити».
Клинические подробности
Клиническое исследование SELECT (NCT03574597) фазы III (рандомизированное, двойное слепое, плацебо-контролируемое, многоцентровое, международное) проверило «Вегови» (Wegovy, семаглутид) или плацебо среди пациентов (n=17604) в возрасте 45 лет и старше с лишним весом или ожирением.
В анамнезе участников должно было быть сердечно-сосудистое заболевание, что подтверждалось хотя бы одним из следующих состояний:
перенесенный инфаркт миокарда;
перенесенный ишемический или геморрагический инсульт;
наличие симптоматической болезни периферических артерий, о которой свидетельствовала перемежающаяся хромота с лодыжечно-плечевым индексом в покое менее 0,85, пройденная процедура реваскуляризации периферических артерий или ампутация по причине атеросклеротического заболевания.
Композитная первичная конечная точка эффективности лечения была установлена временем до первого столкновения с серьезным сердечно-сосудистым событием (MACE), таким как сердечно-сосудистая смерть, нелетальный инфаркт миокарда или нелетальный инсульт.
После наблюдений в течение усредненных (39,8 ± 9,4) месяца, то есть в период приблизительно от 2,5 лет до 4 лет, события MACE были зарегистрированы для 6,5% и 8,0% пациентов в группах семаглутида (semaglutide) и плацебо.
Применение «Вегови» снизило риск MACE на относительных 20%: отношение риска (hazard ratio, HR) 0,80 (95% [здесь и далее]: 0,72–0,90; p<0,001).
Если говорить об отдельных компонентах MACE, назначение семаглутида привело к следующим снижениям рисков относительно плацебо:
сердечно-сосудистая смерть: на 15% (HR 0,85 [0,71–1,01]);
нелетальный инфаркт миокарда: на 28% (HR 0,72 [0,61–0,85]);
нелетальный инсульт: на 7% (HR 0,93 [0,74–1,15]).
Желудочно-кишечные расстройства, такие как тошнота, рвота и диарея, — наиболее распространенные нежелательные явления, которые привели к прекращению лечения семаглутидом.
Экспертные комментарии
Согласно прогнозам, к 2035 году у более половины населения планеты будет избыточная масса тела или ожирения [1]. В 2015 году высокий индекс массы тела (ИМТ) стал причиной 4 млн смертей, свыше двух третей из которых были вызваны сердечно-сосудистыми заболеваниями [2]. Лишний вес и ожирение ассоциированы с повышенным риском серьезных сердечно-сосудистых событий (MACE), причем даже после учета влияния метаболических факторов сердечно-сосудистого риска ввиду избыточного веса [3] [4] [5] [6].
Хотя снижение риска сердечно-сосудистых заболеваний путем лечения дислипидемии [7], гипертонии [8] и сахарного диабета [9] [10] является стандартной доказательной практикой, концепция лечения ожирения с целью снижения риска MACE сдерживается отсутствием должного набора клинических данных, указывающих на то, что модификация образа жизни или фармакологические вмешательства при избыточной массе тела или ожирении улучшают сердечно-сосудистые исходы [11] [12] [13] [14] [15].
Механизмы снижения сердечно-сосудистого риска при применении семаглутида (semaglutide) объясняются физиологическими преимуществами, которые получает организм после уменьшения количества избыточного жира
Во-первых, снижение веса приводит к улучшению уровня глюкозы и ослаблению традиционных факторов промежуточного риска сердечно-сосудистых заболеваний [16].
Во-вторых, уменьшение эктопических отложений жировой ткани благотворно сказывается на сдерживании прогрессирования атеросклероза и дисфункции миокарда [17], притом что периваскулярная и эпикардиальная жировая ткань оказывает прямое неблагоприятное воздействие на сосудистый эндотелий и миокард [18].
В-третьих, избавление от лишнего жира в организме улучшает системную провоспалительную и протромботическую картину, ассоциированную с ожирением [19].
Коррекция избыточной массы тела путем интенсивного изменения образа жизни (путем снижения калорийности рациона и усиленной физической активности) в целом не приводит к улучшению сердечно-сосудистых исходов [11] [14]. Связано это, возможно, с тем, что необходимо добиться снижения веса как минимум на 10% [20], а подобное весьма трудно достижимо без фармакологической поддержки. Напротив, бариатрическая хирургия, предполагающая похудение не менее чем на 20%, обеспечивает существенное снижение частоты MACE [21] [22].
Назначение семаглутида, изученное в этом клиническом испытании, помогло снизить вес в среднем на 9,4%, то есть не перешагнуло необходимый 10-процентный порог. Тем не менее семаглутид смог улучшить сердечно-сосудистые исходы. Есть мнение, что механизмы семаглутида, защищающие сердечно-сосудистую систему, привлекают множество взаимосвязанных путей, в том числе оказывающих последовательное влияние на кардиометаболические факторы риска.
Так, препараты класса агонистов рецептора глюкагоноподобного пептида-1 (GLP1RA), к которым относится семаглутид, в исследованиях на животных с сахарным диабетом и без него улучшили функции эндотелия и левого желудочка, способствовали стабильности атеросклеротических бляшек, снизили агрегацию тромбоцитов [23].
В этом клиническом испытании назначение семаглутида отразилось положительным изменением множества хорошо изученных биомаркеров сердечно-сосудистого риска, таких как артериальное давление, окружность талии, гликемический контроль, нефропатия, уровни липидов и C-реактивного белка. Что примечательно, указанные изменения были достигнуты на фоне применения статиновой терапии, гипотензивных лекарственных средств и прочих препаратов, используемых в лечении атеросклеротического сердечно-сосудистого заболевания.
Следует понимать, что в испытание были включены только пациенты с сердечно-сосудистыми заболеваниями, то есть остается неизвестным влияние семаглутида на первичную профилактику сердечно-сосудистых событий у лиц с избыточной массой тела или ожирением, но без атеросклеротического сердечно-сосудистого заболевания.
Ранее семаглутид подтвердил свою способность снижать риск MACE среди пациентов с сахарным диабетом 2-го типа при наличии сердечно-сосудистого заболевания или сердечно-сосудистых факторов риска: на 26% и 21% — соответственно при назначении семаглутида подкожными инъекциями еженедельно и перорально ежедневно [24] [25]. В этой популяции пациентов, согласно метаанализу, препараты GLP1RA-класса снижают риск MACE на 14% [10].
«Амтагви» (Amtagvi, лифилейцел) — новый лекарственный препарат, предназначенный для лечения неоперабельной или метастатической меланомы у взрослых, ранее прошедших терапию блокатором PD-1 и, при наличии мутации BRAF V600, ингибитором BRAF (вкупе с ингибитором MEK или без него).
«Амтагви», который вводится однократно, одобрен Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) в середине февраля 2024 года.
Регуляторный вердикт вынесен в условном порядке: препарату предстоит окончательно подтвердить свою терапевтическую эффективность.
При данном показании, когда меланома прогрессировала во время или после назначения блокаторов PD-(L)1 и таргетных лекарственных средств, консенсусно утвержденных протоколов лечения не существует. «Амтагви» призван восполнить этот пробел.
Препарат лифилейцел (lifileucel), разработанный «Айованс байотерапьютикс» (Iovance Biotherapeutics), представляет собой адоптивную T-клеточную терапию с использованием инфильтрирующих опухоль лимфоцитов (TIL), поликлональных и индивидуализированных под неоантигенную опухолевую специфику заболевания конкретного пациента.
«Амтагви» появился спустя приблизительно 35 лет после того, как в ходе экспериментального лечения был впервые продемонстрирован терапевтический потенциал TIL в борьбе с онкологическими заболеваниями. «Амтагви» в США — первая аутологичная TIL-клеточная терапия для коммерческого применения и первая однократная персонализированная T-клеточная терапия для лечения солидных опухолей.
Поскольку «Амтагви» не является коробочным препаратом, а требует персонализированного производственного процесса, стоимость лечения установлена высокой. Американским пациентам аутологичная клеточная терапия лифилейцел обойдется в 515 тыс. долларов. Цена находится приблизительно на одном уровне со стоимостью CAR-T-лекарств.
В первой половине 2024 года регистрационное досье лифилейцела отправится в адрес Европейского агентства по лекарственным средствам (EMA).
«Амтагви»: механизм действия лифилейцела
В 2020 году меланома была диагностирована оценочно у 325 тыс. человек во всём мире, что повлекло за собой приблизительно 57 тыс. смертельных исходов [1].
Ингибиторы иммунных контрольных точек (ИИКТ) и таргетная терапия произвели революцию в лечении меланомы на поздних стадиях. И всё же значительная часть пациентов не отвечает на лечение, в конечном итоге сталкиваясь с рецидивом, притом что фармакологический арсенал после прогрессирования заболевания весьма ограничен.
Многие пациенты, получающие ИИКТ (одиночные или в комбинации с другими ИИКТ) в рамках первоочередного лечения меланомы, прогрессируют к 12–18 месяцам терапии [2] [3] [4]. Первичная резистентность к ИИКТ отмечается в 40–65% случаев [2] [5] [6], а приобретенная — в 30–40% [6] [7] [8]. Клинической проблемой является возникновение иммуноопосредованных нежелательных явлений (НЯ) с последующим прекращением ИИКТ-терапии [4] [5] [9].
Ингибиторы BRAF/MEK эффективны для лечения распространенной меланомы [10] [11] [12], но соответствующие мутации, на которые можно таргетно воздействовать, обнаруживаются лишь у 35–50% пациентов [10] [13], ответы на терапию зачастую не являются стойкими, а при рецидиве заболевание весьма быстро прогрессирует [10] [11] [14].
Лечение после прогрессирования меланомы на фоне ИИКТ и таргетной терапии ингибиторами BRAF/MEK продемонстрировало ограниченную пользу. Так, частота общего ответа (ORR) на цитотоксическую химиотерапию составила 4–12% [15] [16] [17], а медиана общей выживаемости (OS) вышла к приблизительно 7 месяцам [17].
Повторное применение ИИКТ является распространенной практикой, хотя Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) такой подход не одобрило, а Национальная всеобщая онкологическая сеть США (NCCN) рекомендует сменить класс препаратов, выбрав другой механизм действия, чем был у предыдущего лечения [18].
Повторное назначение ИИКТ в лице блокатора PD-1 и/или блокатора CTLA-4 после предшествовавшего терапевтического провала ИИКТ обеспечило ORR на уровне 8–29% [19] [20] [21] [22] [23] [24], OS в пределах 5–29 месяцев [19] [20] [21] [22] [24] [25], выживаемость без прогресирования (PFS) в диапазоне 3–5 месяцев [19] [20] [21] [22] и ограниченную продолжительность ответа (DoR) [20] [21] [22]. Аналогичным образом более новая комбинация из блокаторов LAG-3 и PD-1 также показала скромный результат ORR в 11,5% в более запущенных случаях меланомы [26].
Беспокойство вызывают рецидивы иммуноопосредованных НЯ после повторного использования ИИКТ [27] [28]: при возобновлении лечения блокатором PD-1 после предшествовавшей терапии сочетанием из блокаторов PD-1 и CTLA-4, которой сопутствовали тяжелые иммуноопосредованные НЯ, последние, протекавшие с любой степенью тяжести, были зарегистрированы у 50% пациентов, и 30% прекратили лечение по их причине [28].
Подводя итоги, существует высокая неудовлетворенная медицинская потребность в новых эффективных и безопасных вариантах лечения меланомы после провала ИИКТ-терапии.
Адоптивная клеточная терапия с использованием инфильтрирующих опухоль лимфоцитов (TIL) продемонстрировала многообещающую противоопухолевую активность у пациентов с распространенными солидными опухолями [29] [30] [31], включая меланому, рефрактерную к ИИКТ и ингибиторам BRAF/MEK [32] [33] [34] [35] [36].
Микроокружение опухоли изобилует иммуносупрессивными метаболитами (например, аденозином, лактатом), цитокинами (трансформирующим фактором роста бета [TGF-β]) и клетками (регуляторными Т-клетками [Treg], опухоль-ассоциированными макрофагами [TAM], супрессорными клетками миелоидного происхождения [MDSC]). Все они в совокупности снижают эффекторные функции раковых неоантиген-специфических Т-клеток CD4+ и CD8+ — инфильтрирующих опухоль лимфоцитов (TIL) [37] [38] [39]. Есть мнение, что без достаточного количества и качества эндогенных TIL вызов противоопухолевого иммунного ответа характеризуется неэффективностью и неустойчивостью.
Изоляция, экспансия ex vivo и активация TIL у пациентов с рефрактерным заболеванием придает новый импульс Т-клеткам, улучшая их фенотипический, функциональный и опухолереактивный профиль [40] [41]. Применение подобного поликлонального TIL-препарата способно преодолеть иммуносупрессивные барьеры и вызвать устойчивую иммуноопосредованную регрессию опухоли [42] [43], при этом минимизируя селективное давление, характерное для стратегий клеточной терапии с использованием одного антигена-мишени [44].
Лифилейцел (lifileucel, LN-144), разработанный «Айованс байотерапьютикс» (Iovance Biotherapeutics), представляет собой однократную аутологичную клеточную терапию, которая обращается к инфильтрирующим опухоль лимфоцитам, извлеченнным из опухолевой ткани пациента, а затем прошедшим через 22-дневный (впоследствии он будет сокращен до 16 дней) централизованный производственный процесс выпуска миллиардов специфических для пациента поликлональных TIL [33] [40].
Инфильтрирующие опухоль лимфоциты представляют собой смесь T-клеток CD8+ и CD4+ [45] с преимущественно эффекторным фенотипом памяти, что связано с цитотоксической функцией [40] [45]. После однократной инфузии лифилейцела TIL мигрируют в опухолевые очаги по всему организму, где распознают и нацеливаются на множество индивидуализированных опухолеассоциированных неоантигенов и опосредуют лизис опухолевых клеток [33].
Перед вливанием лифилейцела пациент проходит процедуру немиелоабляционного кондиционирования (NMA-LD) для подавления иммуносупрессивного опухолевого микроокружения, а после — короткий курс высокодозного интерлейкина 2 (IL-2) для поддержания T-клеточной активности.
Лифилейцел: клиническая проверка
Клиническое исследование C-144-01 (NCT02360579) фазы II (нерандомизированное, открытое, международное) пригласило взрослых пациентов с неоперабельной или метастатической меланомой (на стадии IIIc или IV).
Среди основных требований к участникам: рентгенографически подтвержденное прогрессирование заболевания после как минимум одной линии системной терапии, включавшей применение блокатора PD-1 и, в случае наличия опухолевой мутации BRAF V600, ингибитора BRAF (мононазначением или в комбинации с ингибитором MEK).
Испытуемые, предварительно прошедшие немиелоабляционную лимфодеплецию циклофосфамидом и флударабином, получили одну инфузию лифилейцела (lifileucel) [в дозе 1×109–150×109 клеток], а затем короткий курс болюсного высокодозного интерлейкина 2 (600 тыс. МЕ/кг, каждые 8–12 часов, максимум 6 доз).
По прошествии медианных 18,7 месяца (0,2–34,1) наблюдений за пациентами (n=66) частота общего ответа (ORR) составила 36% (95% ДИ [здесь и далее]: 25–49), включая 3% полных ответов (CR) и 33% частичных ответов (33%). Стабилизация заболевания (SD) была зарегистрирована у 44% пациентов.
Частота контроля заболевания (DCR) вышла к 80% (69–89).
Медиана длительности ответа (DoR) достигнута не была (11,8–NR), ответ на протяжении как минимум 1 года фиксировался у 69% больных (46–84).
Медиана общей выживаемости (OS) получилась равной 17,4 месяца (11,0–NR). Показатель OS в течение хотя бы 1 года среди пациентов со статусом CR/PR или SD составил 92% и 38% соответственно.
По истечении медианных 36,6 месяца наблюдений за этими же пациентами результаты получились следующими:
ORR 35% (24–48), в том числе CR 8% и PR 27%;
SD: 36%;
медиана DoR: не достигнута (1,4+ — 45,0+).
Ответ на терапию лифилейцелом не зависел от возраста пациентов, предшествовавшего применения блокатора CTLA-4, мутационного статуса BRAF, уровня экспрессии PD-L1.
При добавлении еще одной когорты испытуемых (n=87) с последующим анализом клинических исходов в совокупной выборке пациентов (n=153) результаты, зарегистрированные через медианных 27,6 месяца наблюдений после лечения лифилейцела, таковы:
ORR 31% (24–39), в том числе CR 6% и PR 26%;
SD: 46%;
медиана DoR: не достигнута (1,4+ — 45,0+), у 42% длительность ответа была не менее чем 18 месяцев;
медиана OS: 13,9 месяца (10,6–17,8), частота 12-месячной OS — 54% (46–62).
медиана выживаемости без прогрессирования (PFS): 4,1 месяца (2,8–4,4), частота 12-месячной PFS — 28% (21–36).
Продолжительное наблюдение (медианных 48,1 месяца) выявило следующие тенденции в отношении вероятности остаться в живых на протяжении 4 лет после однократного вливания лифилейцела:
все ответившие на лечение пациенты (n=48): 47% (33–61);
пациенты с ранним ответом (n=39), то есть в статусе CR или PR на 42-й день: 48% (32–63);
пациенты с поздним ответом (n=9), то есть в статусе CR или PR после 42-го дня: 42% (11–71);
пациенты с улучшившимся ответом (n=16), то есть у которых при дальнейшем наблюдении изначальный статус SD улучшился до PR либо исходный статус PR углубился до CR: 68% (40–85);
пациенты без улучшившегося ответа (n=32): 37% (21–54).
Лифилейцел также успешно справился с лечением распространенной мукозальной меланомой — редким ее подтипом, весьма плохо реагирующим на иммунотерапию блокатором PD-1, если сравнивать с немукозальной меланомой: ввиду более низкой опухолевой мутационной нагрузки (TMB).
Анализ подгруппы пациентов с прежде леченной мукозальной меланомой (n=12), за которыми наблюдали в течение медианных 35,7 месяца, установил показатель ORR на уровне 50% (21–79), включая CR 8% и PR 42%. Медианы DoR и PFS достигнуты не были NR (12,5–NR) и NR (1,4–NR). Медиана OS составила 19,4 месяца (7,9–NR).
Инструкция по медицинскому применению лекарственного препарата «Амтагви» (Amtagvi, лифилейцела) снабжена «чернорамочным» предупреждением о рисках длительной тяжелой цитопении, тяжелых инфекций, кардиопульмональных и почечных нарушений.
Лифилейцел: что дальше
«Айованс байотерапьютикс» (Iovance Biotherapeutics) продолжает клиническое испытание TILVANCE-301 (NCT05727904) фазы III (рандомизированное, открытое, с активным препаратом сравнения, многоцентровое, международное). Адоптивная клеточная терапия изучается в ходе первоочередного лечения неоперабельной или метастатической меланомы (на стадии IIIC/D или IV): сравниваются исходы назначения комбинации из лифилейцела (lifileucel) и «Китруды» (Keytruda, пембролизумаб), блокатора PD-1 авторства «Мерк и Ко» (Merck & Co.), с применением только последнего. Результаты будут готовы к весне 2028 года.
Это исследование является подтверждающим для полноценного регуляторного одобрения лифилейцела со стороны Управления по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA).
Лифилейцел проверяется при метастатической меланоме с асимптоматическим метастазированием в головной мозг (NCT05640193 фазы I), метастатической увеальной меланоме (NCT05607095 фазы I), в ходе неоадъювантного лечения (в сочетании с пембролизумабом) местнораспространенной или метастатической меланомы (NCT05176470 фазы I).
Параллельно лифилейцел тестируется при других онкологических заболеваниях. Для полноты картины: помимо оригинального лифилейцела (LN-144) изучаются его усовершенствованные версии, такие как LN-145 (производится за 16 дней вместо 22-х) и LN-145-S1 (производится из TIL, отобранных по наличию экспрессии PD-1).
Так, клиническое исследование IOV-COM-202 (NCT03645928) фазы II оценивает применимость адоптивной TIL-клеточной терапии в борьбе против распространенной, рецидивирующей или метастатической плоскоклеточной карциномы органов головы и шеи — в сочетании с пембролизумабом, а также против местнораспространенного или метастатического немелкоклеточного рака легкого (НМРЛ) — мононазначением либо в комбинации с «Опдиво» (Opdivo, ниволумаб), PD-1-блокатором «Бристол-Майерс Сквибб» (Bristol-Myers Squibb) и опциональным «Ервоеем» (Yervoy, ипилимумаб), ее же блокатором CTLA-4.
Клиническое исследование C-145-04 (NCT03108495) фазы II изучает пригодность лифилейцела с опциональным пембролизумабом в лечении рака шейки матки: рецидивирующей, метастатической или персистирующей плоскоклеточной карциномы, аденосквамозной карциномы или аденокарциномы шейки матки, не пригодной для хирургического вмешательства и/или облучения.
В конце декабря 2023 года FDA поставило на паузу опорное клиническое испытание IOV-LUN-202 (NCT04614103) фазы II экспериментального лечения лифилейцелом распространенного (неоперабельного или метастатического) НМРЛ без геномных мутаций EGFR, ROS или ALK, прогрессировавшего во время или после первоочередного лечения ингибиторами иммунных контрольных точек (ИИКТ) и платиносодержащими химиопрепаратами и ранее прошедшего как минимум один курс таргетной терапии в случае наличия какой-либо иной опухолевой мутации. Регулятор выказал обеспокоенность одним смертельным случаем, который мог наступить по-видимому из-за немиелоабляционной лимфодеплеции.
Согласно промежуточному анализу данных, частота общего ответа (ORR) составила 26% (95% ДИ: 10–48), включая 4% полных ответов (CR) и 22% частичных ответов (PR). Медиана длительности ответа (DoR) достигнута не была (1,4+ — 9,7+).
Обнаружились также весомые предпосылки для добавления лифилейцела к пембролизумабу и платиносодержащим химиопрепаратам в рамках терапии первой линии прежде нелеченного НМРЛ без оглядки на статус PD-L1: здесь показатель ORR вышел к внушительным 80%.
«Айованс» разрабатывает IOV-4001 — генетически модифицированный препарат аутологичных TIL, который обладает усиленной противоопухолевой активностью благодаря инактивации гена PDCD1, кодирующего PD-1. Другими словами, речь идет об объединении в одном лекарстве TIL с блокатором PD-1. Для этого у «Селлектис» (Cellectis) была лицензирована технология генного редактирования на базе подобных активаторам транскрипции эффекторных нуклеаз (TALEN). Клиническое исследование IOV-GM1-201 (NCT05361174) фазы I/II пробует лечить неоперабельную или метастатическую меланому либо распространенный НМРЛ.
«Амтагви»: из истории разработки лифилейцела
В конце мая 2023 года Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) приняло регистрационное досье лифилейцела (lifileucel), вердикт по которому должен был быть вынесен в приоритетном порядке за шесть месяцев — до конца ноября.
В середине сентября 2023 года FDA уведомило о продлении рассмотрения заявки до конца февраля 2024 года — ввиду ограниченных ресурсов американского регулятора.
Изначально «Айованс байотерапьютикс» (Iovance Biotherapeutics) предполагала подать заявку на регистрацию лифилейцела еще во второй половине 2020 года, но не смогла договориться с регулятором об оценочных критериях эффективности клеточной терапии. По итогам было решено перенести отправку досье на первую половину 2022 года. Однако процесс начался лишь в конце августа.
В списке предполагаемых брендовых названий адоптивной клеточной терапии лифилейцел присутствуют следующие торговые наименования: «Тилзелиос» (Tilzelios), «Тилванс» (Tilvance), «Иоватил» (Iovatil), «Магнитил» (Magnitil), «Тилвантидж» (Tilvantage), «Индалифо» (Indalifo), «Неомьюн» (Neomune). Возможно, они будут использованы для следующих клеточных препаратов авторства «Айованс».
Базовый протокол клеточной терапии был разработан Стивеном Розенбергом (Steven Rosenberg) из Национального онкологического института США (NCI) еще в 1990-х годах. В августе 2011 года «Айованс», тогда называвшаяся «Лайон байотекнолоджис» (Lion Biotechnologies), подписала с ним соглашение о сотрудничестве в области исследований и разработок (CRADA).
Что касается интерлейкина 2 (IL-2), добавляемого к лифилейцелу, в конце января 2023 года «Айованс» купила у британской «Клиниджен» (Clinigen) мировые права на «Пролейкин» (Proleukin, алдеслейкин), рекомбинантный IL-2. Авансом заплачено 167 млн фунтов, обещано 42 млн фунтов после первого одобрения лифилейцела, плюс двузначное роялти от реализации «Пролейкина».
Лифилейцел: конкурентная обстановка
Разработкой противораковой клеточной терапии на базе инфильтрирующих опухоль лимфоцитов (TIL) занимаются и другие игроки фармотрасли.
Так, «Тёрнстоун байолоджикс» (Turnstone Biologics) обратилась к дифференцированному подходу, который предполагает тщательный отбор наиболее мощных и опехолерактивных аутологичных T-клеток в целях улучшения клинических исходов лечения солидных опухолей (неоперабельных или рефрактерных к стандартной терапии), таких как рак молочной железы, колоректальный рак, кожная и увеальная меланома, немелкоклеточный рак легкого (НМРЛ), рак органов головы и шеи.
Вдобавок к TIL-терапии «Тёрнстоун» предлагает использовать онколитические вирусы, превращающие иммунологически невосприимчивое «холодное» микрокружение опухоли в более реактивное «горячее».
Британская «Акилиз терапьютикс» (Achilles Therapeutics) пробует усовершенствовать TIL-клеточную терапию путем отбора только клональных неоантиген-реактивных T-клеток (cNeT), которые активнее обычных TIL. Подход изучается в лечении неоперабельных НМРЛ и меланомы.
«Обсидиан терапьютикс» (Obsidian Therapeutics) поставила на TIL-терапию без необходимости в поддерживающей добавке в лице высокодозного интерлейкина 2 (IL-2). Для этого TIL генетически модифицируются так, чтобы они могли продуцировать интерлейкин 15 (IL-15), причем мембраносвязанный, а не секретируемый. Среди преимуществ IL-15 перед IL-2: стимуляция антигенонезависимой экспансии и персистенции TIL, подпитывание экспансии и активности близлежащих естественных киллеров (NK), сдвиг фенотипа в сторону T-клеток CD8+ и T-клеток памяти. IL-15 в отличие от IL-2 подавляет индуцированную активацией клеточную смерть, не увеличивает количество иммуносупрессивных T-регуляторных клеток (Treg), не вызывает токсичность, связанную с синдромом повышенной проницаемости капилляров. Концепция тестируется в лечении неоперабельной меланомы.
«Онивайд» (Onivyde, иринотекан в пегилированной липосомальной рецептуре) отныне можно применять для первоочередного лечения метастатической протоковой аденокарциномы поджелудочной железы у взрослых пациентов. Препарат назначается в комбинации с оксалиплатином, фторурацилом и лейковорином (схема NALIRIFOX).
Соответствующее разрешение выдано Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) в середине февраля 2024 года.
Химиотерапевтическое лекарственное сочетание продлевает общую выживаемость и выживаемость без прогрессирования по сравнению с применением наб-паклитаксела с гемцитабином.
Следует понимать, что рак поджелудочной железы по-прежнему остается фактически недосягаемым ни для какого фармакологического лечения, и потому продление жизни при назначении «Онивайда» получилось относительно скромным. Тем не менее любые успехи в лечении этого очень агрессивного онкологического заболевания следует воспринимать с воодушевлением.
«Онивайд» дебютировал в конце октября 2015 года для лечения метастатической аденокарциномы поджелудочной железы, которая прогрессировала после терапии с использованием гемцитабина. Препарат применяется в комбинации с фторурацилом и лейковорином (схема NALIRIFOX).
Французская «Ипсен» (Ipsen) занимается коммерциализацией «Онивайда» в США, «Лаборатории Сервье» (Les Laboratoires Servier) торгует им в остальных странах, кроме Тайваня, где реализация препарата отдана «Фармаэнджин» (PharmaEngine).
«Онивайд»: механизм действия
Протоковая аденокарцинома поджелудочной железы, самый распространенный тип рака поджелудочной железы, остается одной из самых смертоносных злокачественных опухолей: пятилетняя выживаемость при заболевании с отдаленными метастазами составляет всего 3% [1] [2].
В последнее десятилетие две комбинированные химиотерапевтические схемы — квадруплет фторурацила (fluorouracil), лейковорина (leucovorin), иринотекана и оксалиплатина (oxaliplatin) [FOLFIRINOX] и дублет наб-паклитаксела (nab-paclitaxel) и гемцитабина (gemcitabine) — стали стандартом первой линии лечения протоковой аденокарциномы поджелудочной железы [3] [4] [5]. Однако эти схемы никогда не сравнивались напрямую, что оставляет неопределенность в отношении выбора оптимального варианта терапии.
Ингибиторы иммунных контрольных точек (ИИКТ) продемонстрировали лишь частичную пользу (за исключением рака поджелудочной железы с высокой микросателлитной нестабильностью [MSI-H/dMMR]), и несмотря на большой интерес к использованию геномного профилирования для улучшения клинических исходов, относительно небольшое количество пациентов пригодны для молекулярно-таргетированного лечения [6] [7] [8].
Плохой прогноз и решительно недостаточное количество вариантов лечения, которые были бы доступны большинству пациентов с раком поджелудочной железы, подчеркивают необходимость дальнейших исследований для сравнения эффективных и переносимых новых подходов к лечению, а также для максимизации преимуществ цитотоксических химиотерапевтических схем.
Иринотекан (irinotecan, CPT-11), полусинтетический аналог цитотоксического алколоида камптотецина (camptothecin) с противоопухолевой активностью, является хорошо зарекомендовавшим себя лекарственным средством при раке поджелудочной железы [9] [10] [11] [12].
Во время S-фазы клеточного цикла иринотекан избирательно стабилизирует ковалентные комплексы топоизомеразы I — ДНК, ингибируя рециклирование одноцепочечных разрывов ДНК, опосредованных топоизомеразой I, и вызывая летальные двухцепочечные разрывы ДНК, когда комплексы попадают в механизм репликации ДНК. В итоге иринотекан препятствует процессу репликации клеток, сдерживая рост опухоли [13] [14].
В организме иринотекан метаболизируется с образованием активного метаболита SN−38, который по своей активности многократно превосходит исходную молекулу [15].
Химиотерапевтический лекарственный препарат «Онивайд» (Onivyde, MM-398, PEP02) представляет собой изотоническую дисперсию липосом. Каждая из липосом, будучи однослойным сферическим пузырьком диаметром около 110 нм с липидным бислоем, заключает в себе водное пространство, содержащее приблизительно 80 тыс. молекул иринотекана в гелеобразном или осажденном состоянии в виде октасульфатной соли сахарозы. Липосомальная инкапсуляция защищает иринотекан от раннего превращения в активный метаболит (SN-38), а пегилирование предотвращает быстрое выведение липосом из организма. Всё это способствует эффективной доставке химиотерапевтического соединения в цитозоль из эндосомного отдела клетки [22] [23] [24] [20].
Опухоли поджелудочной железы окружены плотной стромой, которая почти непроницаема. Строма регулирует среду вокруг опухоли, стимулирует ее рост и защищает от проникновения лекарственных молекул. Относительное отсутствие большого количества кровеносных сосудов препятствует должному поступлению противораковых препаратов к злокачественным клеткам [16] [21].
Благодаря макромолекулярному липосомальному носителю «Онивайда» указанная проблема частично преодолевается. Липосомы характеризуются преимущественным накоплением в опухолях в результате эффекта повышенной проницаемости и удержания (ERP), — который объясняется аномальной негерметичной сосудистой сетью опухоли, допускающей экстравазацию макромолекул, и дефектным лимфатическим дренажем, который способствует удержанию этих молекул в микроокружении опухоли [17], — тем самым обеспечивая устойчивое высвобождение в месте опухоли, имитирующее метрономическое дозирование. Создается большое депо иринотекана только в опухоли, приводящее к продолжительной экспозиции SN−38 [18] [19] [20].
Фармакокинетические характеристики липосомальной формуляции иринотекана существенно улучшены в сравнении с таковыми у обычного иринотекана. Так, если клиренс иринотекана из плазмы происходит в течение 8 часов, то «Онивайд» остается в циркуляторном русле свыше 50 часов. Клиренс иринотекана из опухоли осуществляется в течение 24 часов, «Онивайда» — 168 часов. В итоге доза «Онивайда» может быть снижена в 5 раз при сохранении такого же уровня экспозиции, как у иринотекана, что приводит к уменьшению токсичного действия последнего [18].
Французская «Ипсен» (Ipsen) завладела «Онивайдом», после того как в начале 2017 года купила его у «Мерримак фармасьютикалс» (Merrimack Pharmaceuticals), испытывающей трудности с бизнесом.
Клинические подробности
Клиническое исследование NAPOLI 3 (NCT04083235) фазы III (рандомизированное, открытое, с группой активного сравнения, многоцентровое, международное) охватило взрослых пациентов (n=770) с прежде нелеченной метастатической протоковой аденокарциномой поджелудочной железы.
Участникам назначали либо пегилированный липосомальный иринотекан на фоне химиотерапевтического режима FOLFOX (фторурацил + лейковорин + оксалиплатин) — схема NALIRIFOX, либо «Абраксан» (Abraxane, наб-паклитаксел) с гемцитабином — до момента прогрессирования заболевания или неприемлемой токсичности.
Первичная конечная точка эффективности лечения метастатической протоковой аденокарциномы поджелудочной железы была установлена общей выживаемостью (OS). Среди вторичных конечных точек: выживаемость без прогрессирования (PFS) и частота общего ответа (ORR).
По прошествии медианных 16,1 месяца (13,4–19,1) наблюдений показатель OS вышел к медианным 11,1 месяца (95% ДИ [здесь и далее]: 10,0–12,1) в группе «Онивайда» — против 9,2 месяца (8,3–10,6) в группе наб-паклитаксела с гемцитабином. Риск смерти снизился на относительных 17%: отношение риска (hazard ratio, HR) 0,83 (0,70–0,99; p=0,036).
Общая выживаемость на протяжении 12 месяцев составила 46% (41–51) против 40% (35–44), в течение 18 месяцев — 26% (21–32) против 19% (15–24).
Показатель PFS получился равным медианных 7,4 месяца (6,0–7,7) — против 5,6 месяца (5,3–5,8). Риск прогрессирования заболевания или смертельного исхода снизился на 31%: HR 0,69 (0,58–0,83; p<0,0001).
Выживаемость без прогрессирования на протяжении 12 месяцев составила 27% (22–33) против 14% (10–19), в течение 18 месяцев — 11% (11–17) против 4% (0,5–12).
Показатель ORR определился на уровне 42% (37–47) — против 36% (31–41). Медианная продолжительность ответа (DoR): 7,3 месяца (5,8–7,6) — против 5,0 месяца (3,8–5,6).
Терапевтическая схема NALIRIFOX характеризовалась известными нежелательными явлениями (НЯ) со стороны отдельных лекарственных компонентов.
Среди наиболее распространенных НЯ тяжелой или жизнеугрожающей степени выраженности, развившихся на фоне лечения: диарея (у 20% пациентов в группе «Онивайда» — против 5% в контрольной группе), гипокалиемия (15% против 4%), нейтропения (14% против 25%), тошнота (12% против 3%), анемия (11% против 17%), снижение количества нейтрофилов (10% против 14%).
По причине НЯ лечение прекратили 25% пациентов, получавших «Онивайд», — против 23% в контрольной группе, снизили дозу 54% против 49%.
Инструкция по медицинскому применению лекарственного препарата «Онивайд» снабжена «чернорамочным» предупреждением о рисках тяжелой, жизнеугрожающей или летальной нейтропении (нейтропеническая лихорадка или сепсис) и тяжелой диареи.
В предшествовавшем клиническом исследовании NCT02551991 фазы I/II первоочередное применение схемы NALIRIFOX для лечения метастатического рака поджелудочной железы вывело OS и PFS к медианным 12,6 месяца (8,7–18,7) и 9,2 месяца (7,7–12,0).
Согласно регистрационному клиническому исследованию MPACT (NCT00844649) фазы III, первоочередное назначение наб-паклитаксела с гемцитабином обеспечило OS и PFS на уровне медианных 8,5 месяца (7,9–9,5) и 5,5 месяца (4,5–5,9). Долгосрочные наблюдения установили, что в живых на протяжении 3 и более лет остались лишь 4% пациентов с метастатическим раком поджелудочной железы.
«Зелсувми» (Zelsuvmi, бердазимер) — новый лекарственный препарат, предназначенный для лечения контагиозного моллюска у взрослых и детей (в возрасте от одного года).
«Зелсувми», сделанный в виде 10,3-процентного геля, наносится на пораженные участки ежедневно — на протяжении максимум 12 недель. Перед нанесением лекарства его следует приготовить, смешав содержимое двух комплектных тюбиков: в одном находится бердазимер, в другом — вспомогательный гидрогель.
Бердазимер (berdazimer), продвигаемый «Лиганд фармасьютикалс» (Ligand Pharmaceuticals), одобрен Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) в начале января 2024 года.
Кантаридин поможет избавиться от распространенных вирусных поражений кожи.
«Зелсувми»: механизм действия бердазимера
Оксид азота (II) располагает такими терапевтически полезными свойствами, как иммунная модуляция короткого действия и прямая противомикробная активность широкого спектра действия, — всё это обеспечивает местный иммунитет против чужеродных организмов [1].
У оксида азота имеются регуляторные функции, влияющие на NF-κB, иммуномодуляцию, воспаление, выработку цитокинов и апоптоз — вероятно, через S-нитрозилирование белков [2].
Оксид азота также обладает цитотоксическими функциями, которые влияют на репликацию вирусов посредством реактивных молекул кислорода и/или азота [1].
Таким образом, местное применение оксида азота несет с собой примечательный терапевтический потенциал. Однако невозможность хранить и безопасно доставлять стабильную форму оксида азота к месту инфекции или воспаления ограничивает развитие подобного способа лечения [3].
Бердазимер (berdazimer, SB206, NVN1000) представляет собой топический лекарственный препарат, сделанный по фирменной технологии Nitricil, реализованный в виде геля и высвобождающий оксид азота.
Для высвобождения оксида азота и непосредственно перед применением следует смешать два компонента: бердазимер натрия и гидрогель. Последний способствует высвобождению оксида азота из макромолекулы первого, которая состоит из полисилоксановой полимерной основы с ковалентно связанными N-диазениумдиолатами, являющимися донорами оксида азота. Оксид азота стабильно высвобождается и целенаправленно поступает в кожу, что сводит к минимуму системное воздействие [1].
Механизм действия бердазимера против вируса контагиозного моллюска связан, скорее всего, с нитрозилированием белков и модуляцией NF-κB [1] [2] [3] [4].
Бердазимер разработан «Нован» (Novan), которая в середине июля 2023 года обанкротилась и в конце сентября того же года продала почти все свои активы «Лиганд фармасьютикалс» (Ligand Pharmaceuticals). Последняя зарабатывает на роялти от реализации лекарственных средств других фармпредприятий: в ее портфеле собрано свыше 85 коммерциализированных препаратов и экспериментальных проектов.
«Зелсувми»: эффективность и безопасность бердазимера
Клинические исследования B-SIMPLE4 (NCT04535531), B-SIMPLE2 (NCT03927703) и B-SIMPLE1 (NCT03927716) фазы III (рандомизированные, двойные слепые, плацебо-контролируемые, многоцентровые) пригласили пациентов (n=891, 355, 352) в возрасте 6 месяцев и старше, страдающих контагиозным моллюском (число поражений от 3 до 70).
Среди основных характеристик участников: 96% были в возрасте 2–17 лет, 51% мужчин, среднее число моллюсковых поражений — 20.
Испытуемым назначали плацебо или бердазимер — их следовало наносить ежедневно на протяжении максимум 12 недель.
К первичной конечной точке эффективности лечения, заявленной полным устранением всех очагов поражения контагиозным моллюском по прошествии 12 недель, вышли 32%, 30% и 26% участников, получавших бердазимер в B-SIMPLE4, B-SIMPLE2 и B-SIMPLE1, — против 20%, 20% и 22% в группах плацебо.
После 8 недель моллюсковые поражения полностью исчезли у 20% и 14% пациентов в группах бердазимера в B-SIMPLE4 и B-SIMPLE2 — против 12% и 6% в контрольных группах.
Бердазимер характеризовался приемлемой переносимостью. Среди наиболее распространенных нежелательных явлений при назначении бердазимера: болевые ощущения (в том числе жжение и покалывание) у 19% пациентов, покраснение (12%), зуд (6%) — все они носили в основном легко-умеренную степень выраженности.
«Икант» (Ycanth, кантаридин) — новый лекарственный препарат, предназначенный для лечения контагиозного моллюска у пациентов в возрасте от 2 лет.
Кантаридин (cantharidin), сделанный в виде раствора для местного применения, однократно наносится на каждое кожное поражение. По необходимости лечение повторяют через три недели.
«Икант», разработанный «Веррика фармасьютикалс» (Verrica Pharmaceuticals), одобрен Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) в конце июля 2023 года.
Гель бердазимер для аккуратного устранения очагов поражения контагиозным моллюском.
«Икант» — первое лекарственное средство против контагиозного моллюска, утвержденное американским регулятором. До сего момента лечение этой кожной вирусной инфекции осуществлялось в режиме офф-лейбл (вне инструкции).
Со временем «Икант» расширит спектр показаний, подключив лечение обыкновенных бородавок, остроконечных кондилом (аногенитальных бородавок), подошвенных бородавок.
Согласно оценкам отраслевых обозревателей, спрос на «Икант» в 2026 году перешагнет отметку в четверть миллиарда долларов.
«Икант»: механизм действия кантаридина
Кантаридин (cantharidin) — терпеноид и мощный химический везикант (кожно-нарывное вещество), выделенный из жуков-нарывников, к которым относится шпанская мушка (Lytta vesicatoria), с давних времен используемая в качестве афродизиака.
После того как кантаридин поглощен липидными слоями клеточных мембран эпидермиса его механизм действия включает активацию или высвобождение нейтральных сериновых протеаз, разрушающих десмосомные бляшки (клеточные структуры, участвующие в межклеточной адгезии), что приводит к отслоению тонофиламентов (они удерживают клетки вместе). Этот процесс отражается потерей клеточных связей (акантолиз), в конечном итоге вызывая образование внутриэпидермальных волдырей, воспаление и неспецифический лизис кожи, что способствует отслоению инфицированных кератиноцитов и клиренсу вируса контагиозного моллюска [1].
Что примечательно, поражения заживают без образования рубцов, поскольку акантолиз является внутриэпидермальным процессом.
Кантаридин широко используется в клинической практике лечения контагиозного моллюска и бородавок с 1950-х гг. Преимущества кантаридина перед другими методами лечения включают быстрое наступление терапевтического эффекта и отсутствие серьезно выраженных болевых ощущений во время применения [2].
После того как в 1962 году Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) стало запрашивать данные об эффективности уже одобренных лекарственных препаратов, а производители кантаридина их не предоставили, он был изъят из продажи. В 1997 году кантаридин был вновь представлен в качестве нерасфасованной фармацевтической субстанции, что позволило врачам применять его в клинической практике. Однако неоднозначность FDA-статуса кантаридина приводит к ряду проблем, в том числе с его доступностью [3].
Несмотря на повсеместное применение и доказанную безопасность кантаридина, были необходимы надежные клинические испытания, которые окончательно подтвердили бы эффективность и безопасность этого препарата с последующим одобрением его в качестве лекарственного средства против контагиозного моллюска и бородавок.
«Веррика фармасьютикалс» (Verrica Pharmaceuticals) это сделала первой, выпустив топический лекарственный препарат «Икант» (Ycanth), известный под кодовым обозначением VP-102, содержащий 450 мкл раствора с 0,7 об. % кантаридина и реализованный одноразовым аппликатором с 1-мм наконечником. В состав «Иканта» входит краситель генцианвиолет (кристаллический фиолетовый), облегчающий идентификацию уже обработанных и еще не обработанных поражений.
«Икант»: из истории разработки
Появление препарата «Икант» (Ycanth, кантаридин) могло бы состояться существенно раньше, но «Веррика фармасьютикалс» (Verrica Pharmaceuticals) трижды столкнулась с отказом Управления по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA).
Так, в июле 2020 года регулятор отклонил регистрационное досье кантаридина (cantharidin), запросив дополнительную информацию, относящуюся к разделу «Химические свойства, процесс производства и контроль качества» (CMC).
В сентябре 2021 года FDA выявило недостатки на площадке «Стерлинг фармасьютикалс сервисиз» (Sterling Pharmaceuticals Services), контрактной производственной организации (CMO), исполняющей заказы на выпуск «Иканта». Недочеты не были напрямую связаны с кантаридином.
В мае 2022 года FDA отвергло регистрационную заявку «Веррика» ввиду вновь выявленных дефектов на площадках «Стерлинг». Обнаруженные изъяны в основном были связаны с отдельными операциями со стерильной продукцией, относящимися к обслуживанию других заказчиков, притом что производство «Иканта» не требует стерильности. Тем не менее инспекционные правила FDA не позволяют до момента разрешения любых открытых вопросов с производством одобрять какие-либо лекарственные средства, выпускаемые на мощностях проблемного CMO.
«Икант»: эффективность и безопасность кантаридина в лечении контагиозного моллюска
Одинаковые по дизайну опорные клинические исследования CAMP-1 (NCT03377790) и CAMP-2 (NCT03377803) фазы III (рандомизированные, двойные слепые, плацебо-контролируемые, многоцентровые) охватили пациентов (n=528) в возрасте 2 лет и старше с контагиозным моллюском.
Возраст участников в основном укладывался в пределы 2–11 лет, мужчин и женщин было поровну, период от постановки диагноза до начала экспериментального лечения составил в среднем 4,2 месяца, приблизительно треть пациентов ранее проходили лечение контагиозного моллюска, исходное число очагов поражений — в среднем 21,5.
Испытуемым назначали плацебо или кантаридин, которые наносили на все очаги поражения и оставляли на 24-часовой срок. Процедуру повторяли до 4 раз с интервалом в 21 день.
К первичной конечной точке эффективности лечения контагиозного моллюска, установленной пропорцией пациентов с полным исчезновением всех поддающихся лечению поражений (имевшихся и новых [здесь и далее]) на 84-й день, вышли 46% и 54% получавших «Икант» участников CAMP-1 и CAMP-2 — против 18% и 13% в группах плацебо (p<0,0001).
По окончании лечения число поражений уменьшилось в среднем на 69% и 83% — против его роста на 20% и 19% (p<0,05).
Применение «Иканта» отразилось быстрым наступлением терапевтического эффекта: статистически значимое расхождение с группой плацебо наблюдалось уже после первого курса лечения, продолжая улучшаться при последующем применении кантаридина.
При объединении результатов клинических испытаний первичная конечная точка была зафиксирована для 50% пациентов, лечивших контагиозный моллюск кантаридином, — против 16%, проходивших терапию плацебо (p<0,0001). По завершении лечения число поражений уменьшилось в среднем на 76% — против 0,3% (p<0,0001).
Если рассматривать излечение контагиозного моллюска в контексте локализации поражений, результаты следующие: поражения полностью исчезли на голове/шее у 82% получавших «Икант» и у 40% получавших плацебо, на спине/ягодицах — у 75% против 37%, на груди/животе — у 71% против 37%, в паху — у 86% против 52%, на верхних конечностях — у 67% против 34%, на нижних конечностях — у 64% против 33%.
Профиль безопасности «Иканта» характеризовался приемлемой переносимостью: лечение полностью завершили 94% пациентов — против 95% в контрольных группах. Среди наиболее распространенных нежелательных явлений, возникших в ходе лечения, были побочные реакции по месту применения: везикулы, зуд, боль, эритема, струпья, депигментация, сухость. Все они носили в основном легко-умеренную степень выраженности.
Так, клиническое исследование COVE-1 (NCT03487549) фазы II проверило кантаридин в лечении обыкновенных бородавок. Применение препарата привело к полному исчезновению кожных поражений у 52% пациентов.
Клиническое исследование CARE-1 (NCT03981822) фазы II оценило кантаридин в задаче лечения остроконечных кондилом. Назначение препарата обеспечило полное исчезновение кожных поражений у 35% испытуемых, число остроконечных кондилом сократилось на 77%.
Продолжается клиническое исследование NCT05188729 фазы II, проверяющее гипотезу, что внутриопухолевое введение кантаридина поможет в ходе лечения базально-клеточной карциномы.