РЕЗЮМЕ
- Во всём мире рак мочевого пузыря занимает десятое место среди наиболее часто диагностируемых видов онкологических заболеваний.
- В развитых странах рак мочевого пузыря в подавляющем большинстве случаев характеризуется уротелиальной гистологией (ранее относился к переходно-клеточному раку).
- Уротелиальная карцинома мочевого пузыря в три четверти случаев классифицируется как немышечно-инвазивный рак мочевого пузыря (НМИРМП), то есть как рак мочевого пузыря без прорастания в мышечный слой.
- После трансуретральной резекции (ТУР) всей видимой опухоли, как первоочередного хирургического лечения НМИРМП, с последующей поддерживающей иммунотерапией бациллой Кальметта — Герена (БЦЖ), которая весьма эффективна, многие пациенты всё равно сталкиваются с рецидивом или прогрессированием заболевания.
- Существует необходимость в новых способах лечения, дополняющих ТУР и БЦЖ-вакцину и преследующих цель сдерживания заболевания.
ЧТО ПРОИЗОШЛО
«Си-джи онколоджи» (CG Oncology) разработала кретостимоген гренаденорепвек (cretostimogene grenadenorepvec) — онколитический вирус, предназначенный для лечения высокорискового немышечно-инвазивного рака мочевого пузыря (НМИРМП), который не ответил на стандартную иммунотерапию бациллой Кальметта — Герена (БЦЖ).
Речь идет о лечении, направленном на то, чтобы избавить пациентов от проведения оперативного вмешательства, предполагающего радикальную цистэктомию (хирургическое удаление мочевого пузыря), после которой качество жизни резко ухудшается. Ну или максимально отсрочить эту процедуру.
Кретостимоген гренаденорепвек, который безусловно будет одобрен регуляторами, получит одно из брендовых названий: «Трувезик» (Truvesic) или «Арамира» (Aramira).
ПОЧЕМУ ЭТО ВАЖНО
В три четверти случаев рака мочевого пузыря речь идет о немышечно-инвазивном раке мочевого пузыря (НМИРМП). Первоочередное лечение предполагает трансуретральную резекцию всей видимой опухоли. В целях профилактики рецидива заболевания применяют иммунотерапию бациллой Кальметта — Герена (БЦЖ). В ряде случаев НМИРМП возвращается, и варианты его дальнейшего лечения ограничены, притом что радикальная цистэктомия несет за собой резкое ухудшение качества жизни.
Перспективы: лечение рака мочевого пузыря после БЦЖ. Самый полный обзор в мире
Немышечно-инвазивный рак мочевого пузыря (НМИРМП): варианты лечения в обозримом будущем.
КАК ЭТО РАБОТАЕТ
Кретостимоген гренаденорепвек (cretostimogene grenadenorepvec, CG0070) — онколитический вирус, избирательно реплицирующийся в опухолевых клетках с дефектом сигнального пути белка ретинобластомы (RB). Подобная избирательность реализована благодаря тому, что в последовательность аденовируса серотипа 5 (Ad5) вставлен промотор E2F. Дополнительно присутствует трансген, кодирующий гранулоцитарно-макрофагальный колониестимулирующий фактор (GM-CSF) [1].

Белок RB, будучи опухолевым супрессором, предотвращает чрезмерный рост клеток путем ингибирования прохождения по клеточному циклу, до тех пор пока клетка не будет готова к делению. Когда клетка готова к нему, RB фосфорилируется, тем самым инактивируясь, и клеточному циклу разрешается идти дальше [2]. Сверхэкспрессия RB ассоциирована с повышенным риском прогрессирования и сокращенной выживаемостью [3]. При раке мочевого пузыря его опухолевые клетки почти всегда несут RB-мутации [4].
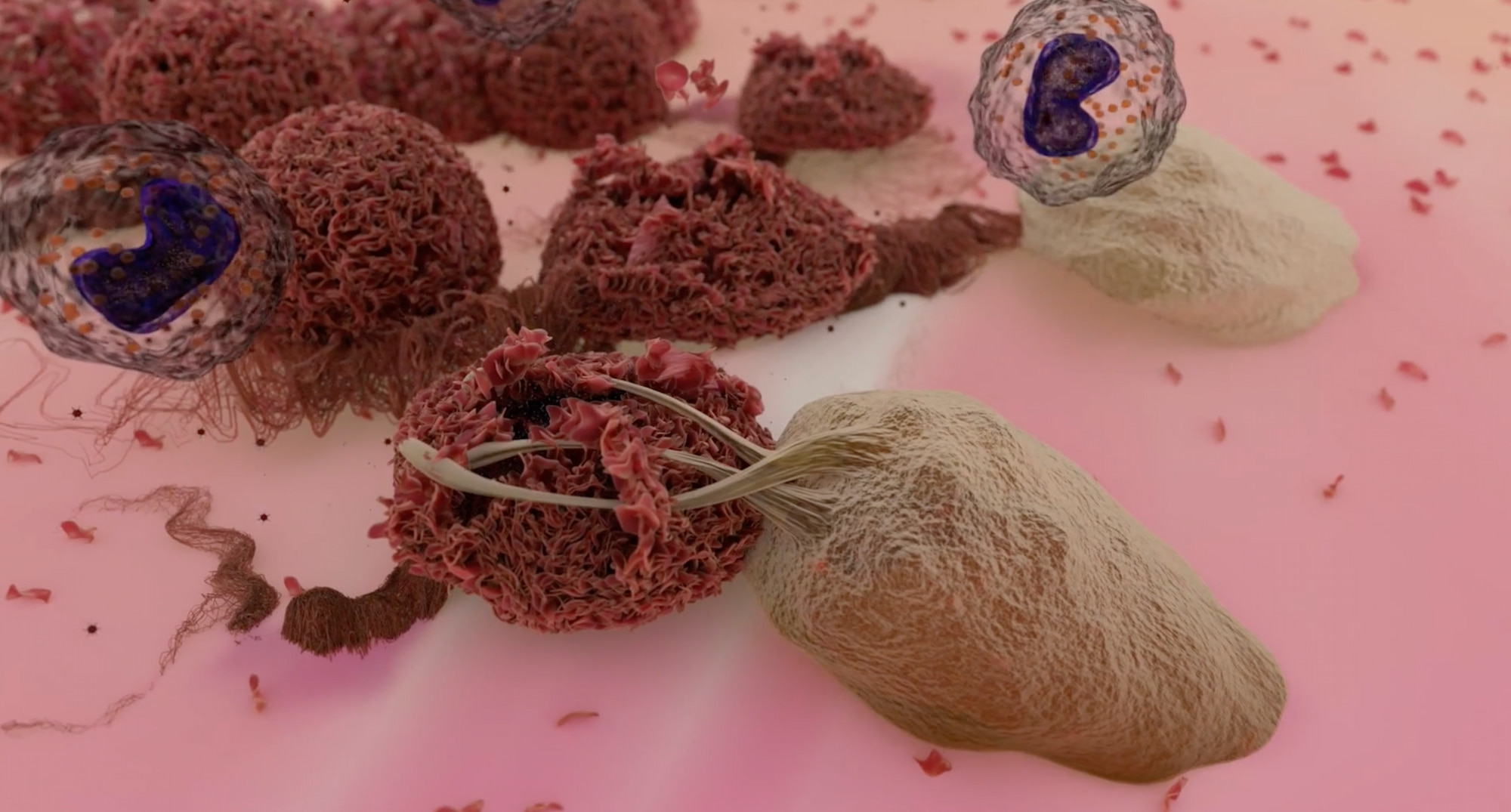
Кретостимоген гренаденорепвек наделен двойным механизмом действия: прямым лизисом опухоли за счет селективной репликации в опухолевых клетках с дефектами сигнального пути RB и иммуноопосредованным их уничтожением в результате иммуногенной гибели опухолевых клеток и локальной выработки GM-CSF — цитокина, критически важного для иммунной активации.
После того как кретостимоген гренаденорепвек инфицировал опухолевые клетки, они начинают погибать, в результате чего высвобождаются неоантигены. GM-CSF запускает дифференцировку антигенпрезентирующих клеток (APC), захватывающих неоантигены, и сдерживает супрессорные клетки миелоидного происхождения (MDSC). Распознавание антигенов на дендритных клетках результирует активацией T-хелперов и цитотоксических лимфоцитов (CTL) наряду с иммунной активацией опухолевого микроокружения (TME) [5].
ЧТО ВЫЯСНИЛОСЬ
«Си-джи онколоджи» (CG Oncology) провела кретостимоген гренаденорепвек (cretostimogene grenadenorepvec) через ряд клинических испытаний, надежно доказавших терапевтическую состоятельность препарата «Трувезик» (Truvesic) / «Арамира» (Aramira).
BOND-003
Продолжающееся клиническое исследование BOND-003 (NCT04452591) фазы III (нерандомизированное, открытое, многоцентровое, международное) пригласило взрослых пациентов (n=112) с высокорисковым раком мочевого пузыря без прорастания в мышечный слой (немышечно-инвазивный), который не отреагировал на ранее проведенную иммунотерапию бациллой Кальметта — Герена (БЦЖ), то есть заболевание персистировало или рецидивировало в течение 12 месяцев по завершении последней.
Среди основных критериев включения в испытание:
- предшествовавшая БЦЖ-терапия предусматривала получение как минимум 5–6 доз первоначального индукционного курса вместе с либо хотя бы 2–3 дозами поддерживающей БЦЖ-терапии, либо хотя бы 2–3 дозами повторного индукционного курса;
- либо только персистирующая или рецидивирующая карцинома in situ (CIS), либо она же с рецидивирующей опухолью Ta или T1 (неинвазивное папиллярное новообразование или с инвазией субэпителиальной соединительной ткани) — должно развиться в течение 12 месяцев после завершения БЦЖ-терапии;
- участники должны были пройти трансуретральную резекцию всех опухолей мочевого пузыря (компоненты Ta и T1);
- непригодность к радикальной цистэктомии или отказ от нее.
Пациентам интравезикально назначали кретостимоген гренаденорепвек (cretostimogene grenadenorepvec) — еженедельно на протяжении первых 6 недель лечения. Если на 13-й недели высокозлокачественное заболевание сохранялось, 6-дозовый цикл повторяли. При отсутствии признаков заболевания (например, полный ответ) переходили к курсу из 3 еженедельных доз. Далее, начиная с 25-й недели, лечение осуществлялось 3 еженедельными дозами каждые 12 недель — до 49-й недели, а затем каждые 24 недели.
Включенные в исследование пациенты с медианой возраста 74 года (43–90) характеризовались весьма запущенным заболеванием: медианное число предшествовавших инстилляций БЦЖ составило 12 (7–66); некоторые также прошли терапию «Китрудой» (Keytruda, пембролизумаб), блокатором PD-1 авторства «Мерк и Ко» (Merck & Co.).
Первичная конечная точка эффективности лечения была установлена пропорцией пациентов, в любое время терапии показавших полный ответ (CR) со стороны CIS (с сопутствующим высокозлокачественным папиллярным новообразованием Ta/T1 или без него).
Согласно предварительным данным, собранным на начало октября 2023 года среди 66 пациентов, за которыми наблюдали в течение не менее чем 3 месяцев, лечение кретостимогеном гренаденорепвеком вывело к CR-статусу 76% больных (95% ДИ [здесь и далее]: 63–85; n=50/66) [1].
3-и 6-месячная частоты CR составили 68% (55–79; n=45/66) и 64% (51–75; n=42/66).
Длительность ответа (DOR) на протяжении как минимум 3 и 6 месяцев оказалась справедливой для 84% (70–92; n=42/50) и 74% (58–86; n=32/43) испытуемых.
Применение кретостимогена гренаденорепвека характеризовалось приемлемой переносимостью. Среди наиболее распространенных нежелательных явлений (НЯ): симптомы со стороны мочеполовой системы, преходящие и носившие легко-умеренную выраженность, такие как спазм мочевого пузыря (у 21% пациентов), поллакиурия (16%), дизурия (14%), императивные позывы к мочеиспусканию (12%), гематурия (11%).
Согласно обновленным на начало апреля 2024 года данным, показатель CR составил 75% (65–83; n=79/105) [2].
Назначение кретостимогена гренаденорепвека отметилось стойкостью терапевтического ответа в течение продолжительного времени. Так, 54% пациентов, прошедших повторный индукционный курс, перешли в CR-статус.
Ответ длительностью 12 месяцев и дольше показали 83% (n=29/35) пациентов. Медиана DOR не достигнута.
В период наблюдений на протяжении 1 года 92% пациентов не нуждались в радикальной цистэктомии. Ни одному из испытуемых в CR-статусе не потребовалась радикальная цистэктомия: у них не обнаруживалось ни узлового, ни метастатического прогрессирования.
12-месячная выживаемость без прогрессирования (PFS) составила 97%. Медиана PFS не зафиксирована.
Спектр наиболее распространенных НЯ легко-умеренной тяжести следующий: спазм мочевого пузыря (у 23% пациентов), поллакиурия (20%), дизурия (15%), императивные позывы к мочеиспусканию (15%), гематурия (14%).
Согласно обновленным на конец сентября 2024 года данным, лечение кретостимогеном гренаденорепвеком CR-статус сохранился у 75% пациентов (65–92; n=82/110) [3].
Медиана DOR всё еще не достигнута (как минимум, 14,5 месяца), притом что длительность ответа уже превысила 27 месяцев.
Терапевтическая эффективность впечатляет: вероятность ответа на протяжении 12 месяцев и дольше составила 64% (51–73), на протяжении 24 месяцев и дольше — 57% (43–68), а 12-месячная PFS вышла к 97%, притом что 90% испытуемых не нуждались в цистэктомии в течение этого периода.
Профиль безопасности остался прежним: спазм мочевого пузыря (у 25% пациентов), поллакиурия (21%), императивные позывы к мочеиспусканию (20%), дизурия (15%), гематурия (13%).







CORE-001
Клиническое исследование CORE-001 (NCT04387461) фазы II (нерандомизированное, открытое, многоцентровое, международное) включило взрослых пациентов с высокорисковым раком мочевого пузыря без прорастания в мышечный слой (немышечно-инвазивный), который не отреагировал на предшествовавшую иммунотерапию бациллой Кальметта — Герена (БЦЖ).
Испытуемым назначали внутрипузырно кретостимоген гренаденорепвек (cretostimogene grenadenorepvec) и внутривенно «Китруду» (Keytruda, пембролизумаб), блокатор PD-1 авторства «Мерк и Ко» (Merck & Co.).
Согласно промежуточным данным, собранным на начало марта 2023 года среди пациентов (n=34), наблюдения за которым велись не менее 3 месяцев, комбинированное лечение привело к полному ответу (CR) у 85% участников (n=29/34) [1].
Статус CR на протяжении 6, 9 и 12 месяцев сохранялся соответственно у 82% (n=27/33), 81% (n=25/31) и 68% респондентов (n=17/25).
Согласно итоговым данным (n=35), собранным на начало февраля и середину мая 2024 года, по прошествии 12 месяцев в CR-статусе находились 57% пациентов (n=20/35; 40–73). Наблюдения на протяжении медианных 26,5 месяца не установили медиану длительности ответа (DOR): она превысила 21 месяц. 24-месячная частота CR вышла к 54% (n=19/35; 37–71) [2] [3].
Из тех пациентов, кто достиг статуса CR после 12 месяцев, 95% (n=19/20) сохранили этот статус сроком на дополнительных 12 месяцев.
Оценочно, вероятность выйти к полному ответу в 12- и 24-месячный периоды оказалась равной 77% (58–89) и 70% (49–83).
Никто из испытуемых не прогрессировал до мышечно-инвазивного рака мочевого пузыря или метастатического заболевания: 24-месячная выживаемость без прогрессирования (PFS) составила 100%.
Выживаемость без необходимости в цистэктомии по прошествии 24 месяцев — 80%; она же составила 100% среди пациентов с полным ответом в этот период времени.
ПЕРСПЕКТИВЫ
Будущее онколитического вируса «Трувезик» / «Арамира» (Truvesic / Aramira, кретостимоген гренаденорепвек) рисуется оптимистичным. Достаточно сравнить его клинические результаты с терапевтической эффективностью прямых конкурентов, уже одобренных для лечения немышечно-инвазивного рака мочевого пузыря (НМИРМП) после провала иммунотерапии бациллой Кальметта — Герена (БЦЖ).
Так, если монотерапия «Трувезиком» / «Арамирой» сгенерировала полный ответ (CR) у 75% пациентов, то комбинация из «Анктивы» (Anktiva, ногапендекин альфа инбакицепт) и БЦЖ выдала CR на уровне 62%, моноприменение «Адстиладрина» (Adstiladrin, надофараген фираденовек) — 51%, мононазначение «Китруды» (Keytruda, пембролизумаб) — 41%.
Кретостимоген гренаденорепвек (cretostimogene grenadenorepvec) обеспечил длительность ответа (DOR) на протяжении 12 месяцев и дольше у 83% пациентов, тогда как ногапендекин альфа инбакицепт (nogapendekin alfa inbakicept) в сочетании с БЦЖ — у 58%, а надофараген фираденовек (nadofaragene firadenovec) или пембролизумаб (pembrolizumab) — у 46%.
Более того, добавление пембролизумаба к «Трувезику» / «Арамире» оказалось успешным: 12- и 24-месячные вероятностные частоты CR составили 77% и 70%, а DOR превысила 21 месяц.
Профиль безопасности кретостимогена гренаденорепвека не вызвал каких-либо претензий: не зарегистрировано тяжелых или жизнеугрожающих нежелательных явлений.

«Адстиладрин»: генная терапия рака мочевого пузыря
Надофараген фираденовек позволит избежать радикальной цистэктомии.
ЧТО ДАЛЬШЕ
«Си-джи онколоджи» (CG Oncology) заинтересована в расширении популяции пригодных к назначению онколитического вируса «Трувезик» / «Арамира» (Truvesic / Aramira, кретостимоген гренаденорепвек) пациентов, и потому в конце февраля 2024 года приступила к клиническому испытания PIVOT-006 (NCT06111235) фазы III, которое проверяет препарат в лечении немышечно-инвазивного рака мочевого пузыря (НМИРМП) промежуточного риска — после трансуретральной резекции [1].
В середине сентября 2024 года было запущено клиническое исследование CORE-008 (NCT06567743) фазы II, изучающее кретостимоген гренаденорепвек (cretostimogene grenadenorepvec) при НМИРМП высокого риска среди пациентов, которые либо ранее не проходили иммунотерапию бациллой Кальметта — Герена (БЦЖ), либо прошли ее и заинтересованы в поддерживающей терапии.
Продолжается клиническое испытание NCT04610671 фазы I, тестирующее добавление «Опдиво» (Opdivo, ниволумаб), блокатора PD-1 авторства «Бристол-Майерс Сквибб» (Bristol-Myers Squibb), к «Трувезику» / «Арамире» при мышечно-инвазивном раке мочевого пузыря (МИРМП) в случае непригодности к назначению цисплатина.
БИЗНЕС
«Си-джи онколоджи» (CG Oncology), основанная в 2010 году, собрала инвестиционных денег в размере 318 млн долларов [1].
В начале января 2024 года, будучи на волне положительной позднестадийной проверки экспериментального онколитического вируса кретостимоген гренаденорепвек (cretostimogene grenadenorepvec), «Си-джи» запустила подготовку к процедуре первичного размещения на фондовом рынке (IPO) [2].
«Си-джи» собиралась привлечь сумму в диапазоне 181–209 млн долларов. Добавление имеющихся денежных средств, их эквивалентов и ценных бумаг наделило бы компанию капиталом в размере почти 370 млн долларов. Из них приблизительно 155 млн долларов планировалось пустить на финансирование исследований и разработок [3].
В конце января 2024 года «Си-джи» стала публичной компанией: в ходе IPO удалось реализовать акций на сумму 437 млн долларов [4].
На начало мая 2024 года рыночная стоимость «Си-джи» составляла 2,42 млрд долларов [5].
За оригинальной разработкой кретостимогена гренаденорепвека стоит «Селл дженисис» (Cell Genesys) [6], которая в октябре 2009 году объединилась с «Байосанте фармасьютикалс» (BioSante Pharmaceuticals) [7]. В ноябре 2010 года «Байосанте» продала «Си-джи», тогда называвшейся «Колд дженисис» (Cold Genesys), права на экспериментальное лекарство [8]. В июне 2013 года «Байосанте» провела слияние с «Эй-эн-ай фармасьютикалс» (ANI Pharmaceuticals) [9]. В марте 2024 года «Эй-эн-ай» подала на «Си-джи» в суд с требованием выплачивать роялти от реализации готового препарата [10].
ТЕМ ВРЕМЕНЕМ
Фармотрасль продолжает разрабатывать новые способы лечения немышечно-инвазивного рака мочевого пузыря (НМИРМП) высокого риска, на который не подействовала иммунотерапия бациллой Кальметта — Герена (БЦЖ). Цель прозрачна: предложить консервативное лечение, которое максимально длительно откладывает необходимость в радикальной цистэктомии.
Перспективы: лечение рака мочевого пузыря после БЦЖ. Самый полный обзор в мире
Немышечно-инвазивный рак мочевого пузыря (НМИРМП): варианты лечения в обозримом будущем.