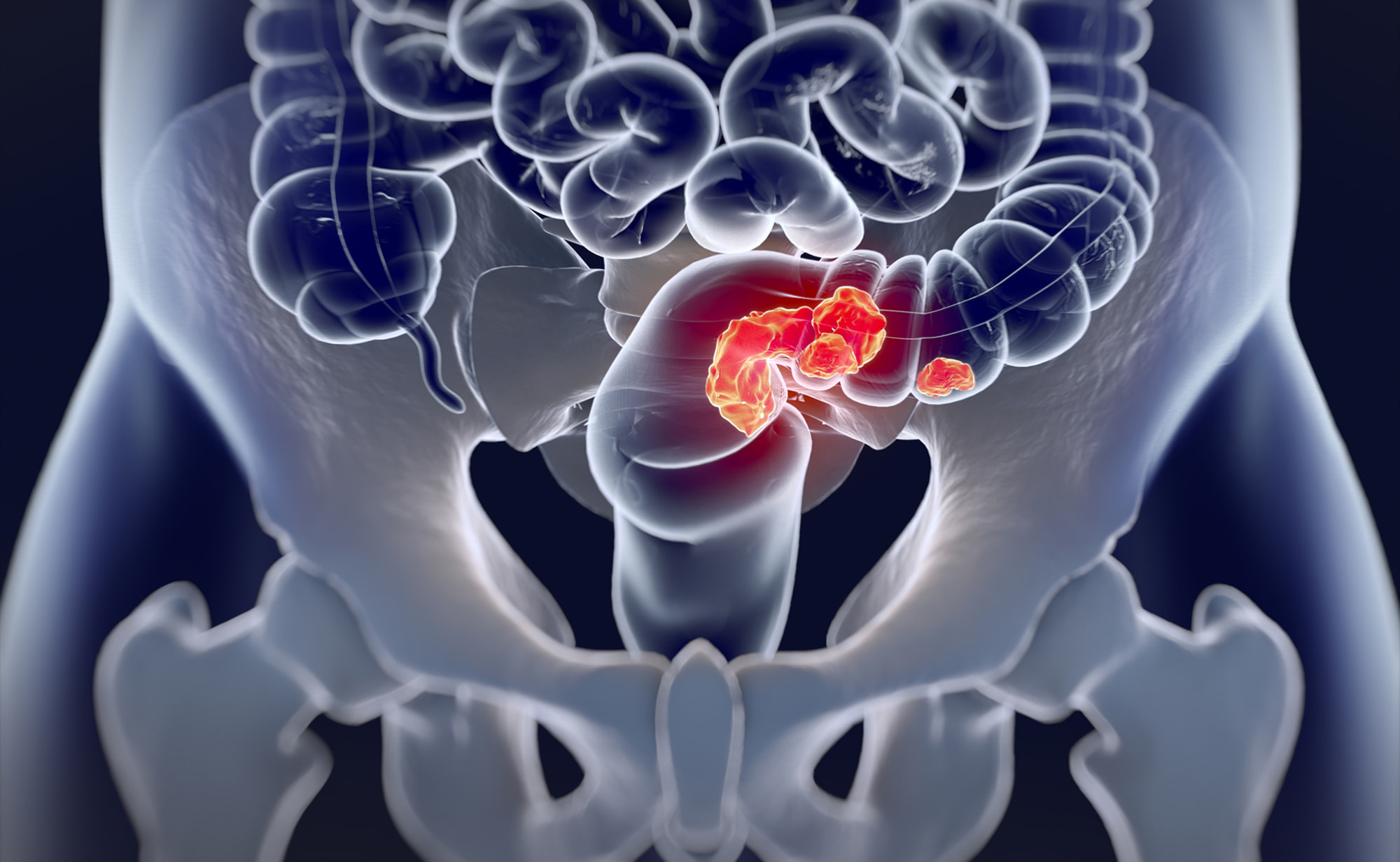
«Джемперли» (Jemperli, достарлимаб) обеспечил прежде невиданную ремиссию в ходе лечения рака прямой кишки.
ОСНОВНЫЕ ФАКТЫ
Шестимесячная монотерапия достарлимабом (dostarlimab) местнораспространенного рака прямой кишки с высокочастотной микросателлитной нестабильностью (MSI-H) или дефектами в системе репарации ошибочно спаренных оснований ДНК (dMMR) привела к тому, что после ее завершения больше половины пациентов, для которых были собраны клинические данные, вышли к стойкой ремиссии, которая продолжалась два года.
Никому из ответивших на экспериментальное лечение испытуемых не потребовалось последующее стандартное лечение.
«Джемперли» (Jemperli, достарлимаб), блокатор PD-1, предлагаемый «ГлаксоСмитКляйн» (GlaxoSmithKline), одобрен в лечении рецидивирующих или распространенных солидных опухолей с dMMR/MSI-H, включая рак эндометрия, идущий по отдельному терапевтическому показанию.
Иммунотерапевтический препарат достарлимаб продлит жизнь при распространенном или рецидивирующем раке эндометрия.
ПРЯМАЯ РЕЧЬ
«Достарлимаб, обеспечивший полную ремиссию на протяжении двух лет, должен стать совершенно новым подходом к лечению местнораспространенного рака прямой кишки с MSI-H/dMMR, не требующего стандартного вмешательства с его изнурительными для пациента последствиями».
Андреа Сёрсек (Andrea Cercek), заведующая отделением колоректального рака и содиректор Центра колоректального и желудочно-кишечного рака с ранним началом Мемориального онкологического центра им. Слоуна — Кеттеринга (MSK, Нью-Йорк, США).
«Собранные клинические данные приближают нас к пониманию потенциала достарлимаба в условиях местнораспространенного рака прямой кишки с MSI-H/dMMR. Беспрецедентная 100-процентная частота клинически полных ответов подтверждает грядущее изменение парадигмы лечения этой формы онкологии».
Хешам Абдулла (Hesham Abdullah), старший вице-президент и руководитель глобальных онкологических исследований и разработок «ГлаксоСмитКляйн» (GlaxoSmithKline).
СУТЬ ВОПРОСА
Рак прямой кишки — это форма рака, которая возникает в конечном отделе толстого кишечника и которая обычно относится к группе онкологических заболеваний, называемых колоректальным раком.
Колоректальный рак занимает третье место в мире по частоте диагностирования [1]. Приблизительно в 5–10% случаев рака прямой кишки отмечаются опухолевые аномалии dMMR/MSI-H, влияющие на корректность восстановления ДНК при ее копировании в клетке [2]. Такие аномалии являются биомаркером, который предсказывает успешный ответ на терапевтическую PD-(L)1-блокаду иммунных контрольных точек [3] [4]. Подобные опухоли чаще всего встречаются при раке эндометрия, колоректальном раке и других желудочно-кишечных онкологических заболеваниях, но могут обнаруживаться и при иных солидных опухолях [5] [6] [7] [8].
ПОЧЕМУ ЭТО ВАЖНО
Нынешний стандарт лечения местнораспространенного рака прямой кишки с MSI-H обращается к первоочередному назначению химиотерапии с облучением, затем проводится опухолевая резекция вместе с участками кишечника и/или окружающих тканей [1]. Это приводит к положительным результатам для большинства пациентов, однако почти одна треть больных в конечном итоге умирает ввиду отдаленного метастазирования [2]. Кроме того, оперативное вмешательство и химиорадиотерапия могут отражаться долгосрочными неблагоприятными последствиями, оказывающими серьезное негативное влияние на качество жизни по причине нарушения функции кишечника и мочеиспускания, половой дисфункции, вторичных раковых заболеваний, бесплодия [1].
КЛИНИЧЕСКИЕ ПОДРОБНОСТИ
Клиническое исследование NCT04165772 фазы II (нерандомизированное, открытое, многоцентровое) привлекло взрослых пациентов (n=16) с прежде нелеченной местнораспространенной ректальной аденокарциномой на стадии II/III с MSI-H/dMMR.
Протокол испытания предполагал, что участники получат неоадъювантный (до хирургического вмешательства) достарлимаб (каждые 3 недели на протяжении 6 месяцев), после чего, в случае остаточного заболевания, будет проведена стандартная лучевая терапия с одновременным назначением химиотерапевтического капецитабина, а затем, в случае сохранения заболевания, — тотальное мезоректальное иссечение.
По прошествии медианных 12 месяцев (6–25) наблюдений за пациентами (n=12), которые прошли лечение достарлимабом, результаты получились следующими [1]:
клинически полный ответ (cCR) составил 100% (95% ДИ [здесь и далее]: 74–100);
опухоль не обнаруживалась никаким из доступных способов, включая МРТ-изображения, ПЭТ-изображения, эндоскопию, пальцевое ректальное исследование, биопсию;
прогрессирования или рецидива заболевания не зафиксировано;
никому из пациентов не потребовались последующие химиорадиотерапия или хирургическое вмешательство.
[su_spoiler class=»my-custom-spoiler» title=»Эволюция эндоскопического и радиографического ответа у пациентов с раком прямой кишки, получавших лечение достарлимабом (dostarlimab). Предупреждение: медицинские изображения могут показаться неприемлемыми.»]
[/su_spoiler]
Столь впечатляющие результаты заставили «ГлаксоСмитКляйн» расширить набор пациентов, чтобы убедиться в высокой эффективности достарлимаба, и она не прогадала.
Так, по прошествии медианных 17,9 месяца (0,3–50,5) наблюдений за испытуемыми (n=42), которые полностью прошли 6-месячный курс лечения при помощи «Джемперли», cCR был зарегистрирован для всех 100% человек [2] [3].
Кроме того, по истечении медианных 26,3 месяца (12,4–50,5) наблюдений, все 24 пациента, для которых был доступен анализ данных, оставались в статусе устойчивого cCR, продолжавшегося как минимум 12 месяцев.
В целом медианное время до фиксирования клинически полного ответа составило 6,22 месяца (6,18–6,45).
ЧТО ДАЛЬШЕ
Продолжается клиническое испытание AZUR-1 (NCT05723562) фазы II, призванное подтвердить успехи достарлимаба в монотерапии прежде нелеченного местнораспространенного рака прямой кишки на стадии II/III (T3–T4, N0 или Tx, N+) с MSI-H/dMMR. Собираются данные, касающиеся пропорции пациентов, сохраняющих статус полной ремиссии по прошествии 12, 24 и 36 месяцев после лечения.
Осуществляется также клиническое исследование AZUR-2 (NCT05855200) фазы III, сравнивающее достарлимаб с химиотерапией в первоочередном лечении колоректального рака на стадии II (T4N0) или стадии III (операбельного) с MSI-H/dMMR. Накапливаются данные по частоте бессобытийной выживаемости (EFS) на протяжении 5 лет после терапии.
ЭКСПЕРТНЫЕ КОММЕНТАРИИ
Победа индукционного неоадъювантного назначения достарлимаба над раком прямой кишки обнадеживает. Облучение и хирургическое вмешательство имеют необратимые последствия для фертильности, сексуального здоровья, функции кишечника и мочевого пузыря [1] [2] [3] [4] [5]. Существенны также последствия для качества жизни, что особенно актуально для молодых людей детородного возраста, поскольку среди них растет заболеваемость раком прямой кишки [6].
Следует, впрочем, понимать, что аденокарцинома прямой кишки только в 5–10% случаев характеризуется наличием MSI-H/dMMR. Такие опухоли весьма плохо отвечают на стандартные схемы химиотерапии, включая неоадъювантную [7] [8] [9].
Важный вопрос заключен в том, почему локализованные опухоли прямой кишки с MSI-H/dMMR реагируют на имммуноонкологическое лечение гораздо сильнее, чем метастатические колоректальные опухоли с MSI-H/dMMR [10].
Одно из объяснений заключается в иммуномодулирующем влиянии микробиома кишечника: некоторые виды бактерий усиливают противоопухолевый иммунный ответ, потенцированный неоадъювантным назначением ингибитора иммунных контрольных точек (ИИКТ) [11] [12] [13]. В ряде клинических испытаний ИИКТ в лечении немелкоклеточного рака легкого (НМРЛ), меланомы и почечно-клеточной карциномы было подтверждено существенное благотворное влияние определенных бактерий кишечного микробиома на противоопухолевый ответ [14] [15] [16].
Кроме того, различия в ответах могут быть связаны с характеристиками опухолевых клеток помимо чрезвычайно высокого мутационного бремени (ввиду нарушенной системы репарации), такими как клональность, анеуплоидия и класс мутации [17] [18] [19].
Если разбивать медианное время до регистрации клинически полного ответа, согласно МРТ прямой кишки, эндоскопии, биопсии, уровню циркулирующей опухолевой ДНК (ctDNA) или ПЭТ-КТ, то оно получилось соответственно равным 6,15 месяца (6,09–6,25), 6,18 месяца (3,62–6,22), 1,41 месяца (1,38–2,73), 1,38 месяца (1,38–2,76) и 2,76 месяца (2,73–6,18).
Получается, что наиболее прогностически ранним оказался высокочувствительный анализ на ctDNA: изначально она обнаруживалась у 97% пациентов, а уже через 6 недель лечения перестала выявляться у более половины участников.
Продолжение наблюдений за пациентами из NCT04165772, чтобы установить окончательную продолжительность ремиссии, позволит выяснить, избавляет ли неоадъюватное иммуноонкологическое лечение от необходимости в хирургической резекции в долгосрочной перспективе.
В будущем, возможно, неоадъюватная блокада PD-1 окажется востребованной в лечении других опухолей с MSI-H/dMMR, таких как локализованный рак поджелудочной железы, желудка, предстательной железы. Следует подтвердить гипотезу востребованности высокоактивной противораковой терапии в неоадъювантном контексте до химиорадиотерапии и хирургического вмешательства, то есть до воздействия других агентов, которые могут нацелиться на клетки с резистентным фенотипом.
Экспериментальный препарат бепировирсен (bepirovirsen), разрабатываемый «ГлаксоСмитКляйн» (GlaxoSmithKline), располагает необходимым потенциалом, для того чтобы обеспечить функциональное излечение хронического вирусного гепатита B.
ОСНОВНЫЕ ФАКТЫ
Продемонстрировано, что еженедельные подкожные инъекции бепировирсена привели к полному клиренсу ДНК и поверхностного антигена вируса гепатита В (HBsAg) у почти трети пациентов после 6 месяцев лечения.
Впрочем, по прошествии 6 месяцев наблюдений после завершения терапии статус функционального излечения оказался справедливым для существенно меньшей пропорции больных. Однако терапию всё равно можно назвать успешной, если сравнивать с существующими лекарственными средствами.
В настоящее время хронический вирусный гепатит B, которым заражены 254 млн человек во всём мире, неизлечим: приходится придерживаться пожизненной терапии, которая далеко не всегда оказывается успешной [1].
Напротив, хронический вирусный гепатит C считается полностью излечимым заболеванием. С мая 2011 года стали планомерно появляться всё более эффективные противовирусные препараты прямого действия (ПППД), за несколько месяцев исцеляющие эту инфекционную болезнь.
Цена лекарств постоянно растет, несмотря на отсутствие значимых улучшений. Разбираемся, почему фармацевтическая индустрия превратилась в машину для выкачивания денег.
КАК ЭТО РАБОТАЕТ
Бепировирсен (bepirovirsen, GSK3228836, IONIS-HBVRx) представляет собой антисмысловой олигонуклеотид (ASO) — одноцепочечную ДНК, которая комплементарна 20 консервативным нуклеотидным последовательностям всех матричных РНК (мРНК) вируса гепатита B (HBV), включая его прегеномную РНК (пгРНК).
Связывание бепировирсена с мРНК и пгРНК HBV приводит к образованию гибридного комплекса, который рекрутирует эндогенную рибонуклеазу H (РНКаза H). Этот фермент расщепляет мРНК и пгРНК HBV, тем самым препятствуя трансляции белков вируса. В результате уменьшается количество РНК, ДНК и белков HBV, в том числе поверхностного антигена вируса гепатита В (HBsAg). Вирусная нагрузка снижается, сдерживаются процессы инфицирования и репликации HBV, появляется шанс на функциональное излечение хронического вирусного гепатита B [1] [2].
Бепировирсен также проявляет агонистическую активность в отношении толл-подобного рецептора 8 (TLR8), тем самым работая как иммуностимулятор, индуцирующий активность врожденного иммунитета и выработку цитокинов [3] [4] [5] [6] [7].
Поскольку бепировирсен не конъюгирован с аминосахаром N-ацетилгалактозамином (GalNAc), нужным для более таргетной доставки в печень, он в большей степени распределяется в непаренхимных клетках печени, нежели в гепатоцитах, и потому распознается резидентными иммунными клетками печени, что приводит к активации сигнализации врожденной иммунной системы [8].
Бепировирсен разработан «Айонис фармасьютикалс» (Ionis Pharmaceuticals), которая в конце августа 2019 года лицензировала его «ГлаксоСмитКляйн» (GlaxoSmithKline). Взамен обещано до 262 млн долларов и роялти от реализации готового лекарственного препарата [9].
Состоятельность терапевтической парадигмы антисмысловых олигонуклеотидов подтверждена немалым числом уже одобренных лекарственных препаратов, направленных на сдерживание экспрессии специфических белков, патогенных в случае какого-либо заболевания. Так, например, «Эксондис 51» (Exondys 51, этеплирсен), «Виондис 53» (Vyondys 53, голодирсен), «Амондис 45» (Amondys 45, касимерсен) и «Вилтепсо» (Viltepso, вилтоларсен) применяются в лечении мышечной дистрофии Дюшенна, «Спинраза» (Spinraza, нусинерсен) используется в лечении спинальной мышечной атрофии, «Тегседи» (Tegsedi, инотерсен) назначается для лечения полинейропатии при наследственном транстиретиновом амилоидозе.
КЛИНИЧЕСКИЕ ПОДРОБНОСТИ
В целях предварительного выяснения эффективности и безопасности лечения хронического вирусного гепатита B при помощи бепировирсена «ГлаксоСмитКляйн» положилась на клиническую программу из трех испытаний фазы II:
B-Clear (NCT04449029): 24- или 12-недельное назначение бепировирсена (разными схемами), причем либо на фоне терапии нуклеозидными/нуклеотидными аналогами (NA), либо без нее.
B-Together (NCT04676724): 24- или 12-недельное применение бепировирсена на фоне NA-терапии, за которым следует 24-недельный курс пегилированного интерферона альфа-2a.
Первичная конечная точка эффективности лечения хронического вирусного гепатита B в первых двух клинических испытаниях была установлена пропорцией пациентов, продемонстрировавших устойчивую вирусную супрессию (подавление вирусной нагрузки; SVR), под которой понимают снижение уровней поверхностного антигена вируса гепатита В (HBsAg) и ДНК HBV ниже определяемых высокоточным методом ПЦР порогов (соответственно 0,05 МЕ/мл и 20 МЕ/мл), сохраняющееся на протяжении 24 недель после завершения лечения и при условии отсутствия в этот период дополнительной терапии какими-либо противовирусными препаратами (если таковые ранее не принимались).
B-Sure (NCT04954859): среди ранее прошедших лечение бепировирсеном и ответивших на него пациентов из других клинических испытаний. В ходе 33-месячных наблюдений выясняется, как долго сохраняется SVR-статус.
B-CLEAR
Клиническое исследование B-Clear (NCT04449029) фазы IIb (рандомизированное, двойное слепое, плацебо-контролируемое, многоцентровое, международное) пригласило взрослых пациентов (n=457) с хроническим вирусным гепатитом B, проходящих либо нет фоновое лечение нуклеозидными/нуклеотидными аналогами (NA).
На протяжении 24 недель участникам еженедельно подкожными инъекциями назначали бепировирсен (разными схемами):
группа 1: бепировирсен 300 мг — 24 недели;
группа 2: бепировирсен 300 мг — 12 недель, затем бепировирсен 150 мг — 12 недель;
группа 3: бепировирсен 300 мг — 12 недель, затем плацебо — 12 недель;
группа 4: плацебо — 12 недель, затем бепировирсен 300 мг — 12 недель.
Группы 1, 2 и 3 также получили нагрузочные дозы бепировирсена: по 300 мг на 4-й и 11-й дни.
Согласно промежуточному анализу собранных данных, после 24-недельной терапии хронического вирусного гепатита B наилучшую результативность показала группа 1. Так, одновременное отсутствие HBsAg и ДНК HBV зафиксировано для 28% и 29% испытуемых, соответственно придерживавшихся фоновой терапии NA и не принимавших такие препараты. При этом у 68% и 65% участников уровень HBsAg упал ниже 100 МЕ/мл [1].
Итоговые результаты клинического исследования получились следующими [2].
Среди тех пациентов, которые придерживались фоновой терапии NA, к первичной конечной точке эффективности лечения вышли 9%, 9%, 3% и 0% участников в группах 1, 2, 3 и 4. Среди тех, кто не получал NA, первичная конечная точка достигнута среди 10%, 6%, 1% и 0% испытуемых.
Если допустить «всплески» активности вируса гепатита B (однократное повышение HBsAg или ДНК HBV до уровней, превышающих или равных пороговым), то есть несколько ослабить критерии выхода к первичной конечной точке, ее достигли 10%, 9%, 4% и 2% пациентов на фоновой терапии NA и 14%, 6%, 1% и 4% пациентов без фоновой терапии NA.
Успешный ответ на назначение бепировирсена напрямую зависел от исходного уровня HBsAg до начала лечения: он чаще фиксировался у испытуемых с низким уровнем HBsAg (≤ 3 log10 МЕ/мл) и реже с высоким (> 3 log10 МЕ/мл). К примеру, в группе 1 первичная конечная точка была засвидетельствована у 16% и 25% пациентов с низким изначальным уровнем HBsAg и у 6% и 7% пациентов с высоким — соответственно среди получавших и не получавших фоновую терапию NA.
Бепировирсен характеризовался приемлемой переносимостью. Наиболее распространенным нежелательным явлением были реакции по месту введения препарата.
B-TOGETHER
Клиническое исследование B-Together (NCT04676724) фазы IIb (рандомизированное, открытое, многоцентровое, международное) пригласило взрослых пациентов (n=108) с хроническим вирусным гепатитом B.
Испытуемым, продолжающим следовать стабильной терапии нуклеозидными/нуклеотидными аналогами (NA), назначали бепировирсен (еженедельными подкожными 300-мг инъекциями) на протяжении 24 недель (группа 1) или 12 недель (группа 2); плюс нагрузочные дозы бепировирсена (по 300 мг на 4-й и 11-й дни). Далее участники проходили 24-недельный курс пегилированного интерферона альфа-2a (в еженедельной подкожной дозе 180 мкг).
Экспериментальное лечение обеспечило выход к первичной конечной точке для 9% и 15% пациентов в группах 1 и 2. При этом исходный уровень HBsAg коррелировал с пропорцией ответивших на лечение больных. Так, при низком изначальном уровне HBsAg (≤ 1000 МЕ/мл) первичная конечная точка была зафиксирована для 24% и 41% испытуемых, тогда как при высоком его уровне (≤ 3000 МЕ/мл) — для 14% и 26% [1].
Большинство пациентов (58% в каждой группе), показавших ответ на лечение по завершении применения бепировирсена, не столкнулись с рецидивом инфекции в ходе назначения интерфероновой терапии. Другими словами, добавление интерферона снизило риск рецидива хронического вирусного гепатита B.
Однако только 2 человека (в группе 2) с частичным ответом после бепировирсена вышли к полному ответу в процессе добавления интерферона. То есть подключение последнего к бепировирсену не привело к значимому улучшению исходов, обусловленных снижением уровня HBsAg.
Был отмечен временный рост уровня АЛТ выше утроенной верхней границы нормы. В ходе интерфероновой терапии не было отмечено ассоциации между ростом HBsAg и АЛТ.
Серьезные нежелательные явления (НЯ), связанные с лечением, были зарегистрированы только у 1 пациента (2%). Некоторые НЯ вынудили выйти из исследования 4% испытуемых (n=4): аллергический дерматит, реакции по месту введения препарата, депрессия.
B-SURE
Продолжающееся клиническое испытание B-Sure (NCT04954859) фазы II (нерандомизированное, открытое, многоцентровое, международное) поставило своей целью выяснить, как долго сохраняется SVR-статус среди ранее прошедших лечение бепировирсеном и ответивших на него пациентов с хроническим вирусным гепатитом B из других клинических испытаний этого экспериментального препарата. Наблюдательное исследование продолжается сроком максимум 33 месяца.
Положительный SVR-статус разнесен по критерию ответа на бепировирсен: полный ответ (CR) и частичный ответ (PR). Первый, отражающий функциональное излечение хронического вирусного гепатита B, предполагает снижение уровней поверхностного антигена вируса гепатита В (HBsAg) и ДНК вируса гепатита B (HBV) ниже порогов, определяемых высокоточным ПЦР-методом: соответственно 0,05 МЕ/мл и 20 МЕ/мл. Второй — уровень HBsAg < 100 МЕ/мл и уровень ДНК HBV ниже вышеуказанного порогового.
Если говорить о пациентах из B-Clear (NCT04449029), то анализ исходов B-Sure осуществляется согласно разбивке по факту терапии нуклеозидными/нуклеотидными аналогами (NA): одна группа пациентов из B-Clear получала фоновое NA-лечение, но затем, по прошествии 3 месяцев после начала участия в B-Sure, должна была прекратить, тогда как вторая группа вообще не проходила фоновую NA-терапию.
В группе фонового NA-лечения оказались 11 человек с полным ответом, из которых 9 пациентов впоследствии, согласно протоколу исследования, прекратили прием NA-препаратов. По прошествии 6 месяцев после остановки NA-терапии полный ответ сохранился у 78% (n=7/9) участников. По прошествии еще 6 месяцев (всего 12), когда 1 больной был исключен из анализа ввиду недоступности данных наблюдений за ним, полный ответ сохранился у всех оставшихся 6 человек, то есть составил 67% (n=6/9).
Ситуация с пациентами, показавшими частичный ответ к моменту включения в B-Sure, следующая. Из 29 пациентов прием NA-препаратов должным образом прекратили 23 человека. По истечении 6 месяцев частичный ответ сохранился у 22% испытуемых (n=5/23), притом что 13% (n=3/23) продемонстрировали отложенный полный ответ. После 12 месяцев частичный ответ сохранился у 13% (n=3/23), а отложенный полный ответ — у 13% (n=3/23).
В группу, не проходившую фоновое NA-лечение, попали 16 пациентов: 11 человек с полным ответом и 5 с частичным. По прошествии 15 месяцев полный ответ сохранился у 36% (n=4/11), частичный — у 20% (n=1/5).
Таким образом, лечение хронического вирусного гепатита B при помощи бепировирсена способно привести к функциональному излечению этой инфекции, что подтверждается результатами долгосрочных наблюдений за пациентами, прекратившими всякое лечение заболевания.
ЧТО ДАЛЬШЕ
На волне обнадеживающих результатов, продемонстрированных бепировирсеном в задаче излечения хронического вирусного гепатита B, «ГлаксоСмитКляйн» запустила два идентичных опорных клинических испытания, B-Well 1 (NCT05630807) и B-Well 2 (NCT05630820), фазы III (рандомизированных, двойных слепых, плацебо-контролируемых, многоцентровых, международных), которые, если завершатся успешно, лягут в основу регистрационного досье.
Среди основных требований к взрослым участникам (n=900 и n=900): стабильная терапия нуклеозидными/нуклеотидными аналогами (NA) на протяжении не менее чем 6 месяцев; уровень HBsAg в пределах 100–3000 МЕ/мл; уровень ДНК HBV < 90 МЕ/мл; уровень АЛТ не выше удвоенной верхней границы нормы.
Испытуемые, продолжающие следовать фоновой NA-терапии, вначале проходят 24-недельный курс терапии бепировирсеном или плацебо, а затем на протяжении 24 или 48 недель получают только NA-препараты.
Первичная конечная точка эффективности лечения установлена функциональным излечением хронического вирусного гепатита B, факт которого подтверждается устойчивой вирусной супрессией (SVR) на протяжении хотя бы 24-недельного периода, оставляемого без какого-либо лечения. Исследования завершатся ближе к концу 2025 года.
В клиническом исследовании B-United (NCT06537414) фазы IIb пациентам (n=280) с хроническим вирусным гепатитом B, придерживающимся стабильной NA-терапии и находящимся в SVR-статусе, вначале назначают комбинацию из даплусирана (daplusiran) и томлигисирана (tomligisiran) — по 50 и 200 мг каждые 4 недели на протяжении 24 недель, а затем проводят 24-недельный курс лечения бепировирсеном. Результаты испытания, которое должно выяснить частоту функционального излечения инфекции, будут готовы к концу 2027 года.
Комбинация из даплусирана и томлигисирана (JNJ-3989, JNJ-73763989, GSK5637608, ARO-HBV) — фиксированная доза малых интерферирующих РНК (миРНК), таргетированных на гепатоциты и индуцирующих процесс эндогенной интерференции для расщепления транскриптов РНК HBV, экспрессируемых как из ковалентно замкнутой кольцевой ДНК (кзкДНК) HBV, так и из ДНК HBV, интегрированной в геном хозяина. Это приводит к снижению уровня всех белков HBV (HBsAg, HBeAg) и его прегеномной РНК (пгРНК) [1].
Самостоятельно сочетание даплусирана и томлигисирана снижает уровень HBsAg, причем независимо от его исходной концентрации, но обеспечить функциональное излечение хронического вирусного гепатита B не в силах. Антисмысловой олигонуклеотид бепировирсен продемонстрировал свою максимальную эффективность в отношении устойчивой потери HBsAg среди пациентов с изначально относительно низким уровнем HBsAg (≤ 3000 МЕ/мл). Отсюда и родилась гипотеза, что, если снизить уровень последнего перед назначением бепировирсена, можно увеличить пропорцию пациентов, которые выйдут к статусу функционального излечения [2].
В клиническом испытании B-Focus (NCT06497504) фазы II изучается лечение пациентов (n=150) с коинфекцией хронического вирусного гепатита B и вируса иммунодефицита человека 1 (ВИЧ-1). Последний должен находиться в статусе вирусной супрессии благодаря антиретровирусной терапии (АРТ). Результаты будут собраны к середине 2027 года.
ЧТО ЕЩЕ
«ГлаксоСмитКляйн» осуществляет клиническое исследование NCT05276297 фазы II, в котором пациентам (n=184) после 12- или 24-недельной терапии хронического вирусного гепатита B бепировирсеном следует назначение экспериментальной таргетной иммунотерапии GSK3528869A.
Первичная конечная точка эффективности лечения установлена устойчивой вирусной супрессией (SVR) по прошествии 24 недель после терапии. Испытание должно завершиться к зиме 2026 года.
GSK3528869A представляет собой иммунотерапевтическую вакцину из трех компонентов:
ChAd155-hIi-HBV: лишенный возможности реплицироваться аденовирус шимпанзе группы C серотипа 155, кодирующий последовательности двух белковых антигенов HBV: усеченного ядерного антигена вируса гепатита В (HBcAg) и полноразмерного малого поверхностного антигена вируса гепатита В (S-HBsAg);
MVA-HBV: кодирующий два вышеуказанных белковых антигена HBV модифицированный осповакцинный вирус Ankara (Modified vaccinia Ankara, MVA), представляющий собой высокоаттенуированный штамм вируса осповакцины (Vaccinia virus);
HBc-HBs/AS01B-4: вышеуказанные белковые антигены HBV, подкрепленные адъювантом AS01B-4, который представляет собой липосомальное сочетание 3-О-дезацилированного 4′-монофосфорил-липида A (MPL) сальмонеллы (Salmonella minnesota) и молекулы сапонина (QS-21) из растительного экстракта квиллайи мыльной (Quillaja saponaria).
Первый компонент GSK3528869A вводится по завершении курса бепировирсеном: в 1-й день, второй — в 57-й, третий — в 113-й и 169-й.
Концептуальная идея применения терапевтических вакцин вроде GSK3528869A состоит в том, что недостаточность индукции HBV-специфического B- и T-клеточного иммунитета ответственна за отсутствие полного клиренса вируса гепатита B [1] [2] [3] [4]. Вакцина должна запускать формирование сильного вирусоспецифического иммунитета против антигенов HBV, контролирующего инфекцию путем индукции нейтрализующих антител и элиминации инфицированных гепатоцитов при участии эффекторных T-клеток [4] [5].
В начале декабря 2024 года «ГлаксоСмитКляйн» остановила клиническую проверку NCT03866187 фазы I/II иммунотерапевтической вакцины GSK3528869A, назначаемой пациентам (n=135) с хроническим вирусным гепатитом B, находящимся в SVR-статусе благодаря терапии нуклеозидными/нуклеотидными аналогами (NA). Заявлено об отсутствии должной эффективности по прошествии 24 недель после лечения [6].
Клиническое исследование NCT05330455 фазы I/II, которое должно завершиться к концу 2027 года, тестирует среди пациентов (n=132) с хроническим вирусным гепатитом B сочетание из бепировирсена и GSK3965193, низкомолекулярного ингибитора неканонической атипичной поли(А)-полимеразы 5 и 7 (PAPD5 и PAPD7). Эти ферменты нужны для стабилизации РНК HBV посредством вирусного посттранскрипционного регуляторного элемента (PRE) [7] [8] [9]. Ингибирование PAPD5 и PAPD7 приводит к подавлению вирусной репликации и синтеза вирусных белков, включая HBsAg [10] [11] [12]. В доклинических исследованиях на мышиной модели HBV продемонстрирована оправданность комбинации бепировирсена и GSK3965193 с позиции усиления снижения уровня HBsAg [13].
ЭКСПЕРТНЫЕ КОММЕНТАРИИ
Все существующие стратегии лечения хронического вирусного гепатита B преследуют цель долгосрочной супрессии (подавления) уровня ДНК вируса гепатита B (HBV). При этом весьма желательной является потеря антигена e вируса гепатита B (HBeAg) у HBeAg-положительных пациентов, поскольку она отражает наличие частичного иммунного контроля над инфекцией. В качестве дополнительной цели следует рассматривать нормализацию уровня АЛТ.
Оптимальной конечной точкой лечения выступает устойчивая потеря HBsAg, так как она указывает на глубокую супрессию репликации HBV и экспрессии вирусного белка, свидетельствуя о функциональном излечении (вирусная супрессия на протяжении не менее чем 6 месяцев) хронического вирусного гепатита B, то есть когда вирус не полностью элиминирован (устранен) из организма, но иммунная система контролирует его без каких-либо лекарственных препаратов [1] [2].
Доступные медикаментозные подходы к лечению хронического вирусного гепатита B, представленные пэгинтерфероновой терапией и назначением нуклеозидных/нуклеотидных аналогов (NA), не могут похвастаться безоговорочной эффективностью. Так, потеря HBsAg происходит весьма редко: по прошествии 6 месяцев после годичного курса лечения это наблюдается в 3–7% случаев пэгинтерфероновой терапии и 0–3% случаев терапии NA. Если применение NA продолжается долго, скажем, 5–8 лет, вероятность потери HBsAg повышается, но опять же незначительно: до 10–12% у изначально HBeAg-положительных пациентов и до менее чем 1–2% у HBeAg-отрицательных [1].
Столь скромная эффективность лечения хронического вирусного гепатита B обусловлена тем, что полная эрадикация HBV нынешними препаратами затруднена по причине сохранения в гепатоцитах как ковалентно замкнутой кольцевой ДНК (кзкДНК) HBV, так и интегрированной в их ядро ДНК HBV, являющихся транскрипционными шаблонами для возобновления репликации ДНК HBV [3] [4].
Попытки комбинированного лечения обеспечили потерю HBsAg в 14% случаев, если после минимум 48-недельной NA-терапии переключить пациентов на пэгинтерфероновую терапию [5]. Считается, что прямая противовирусная активность NA, за счет ингибирования вирусной ДНК-полимеразы (обратной транскриптазы) приводящая к вирусологической супрессии и подавлению репликации HBV, частично восстанавливает адаптивный иммунитет, тем самым способствуя улучшению иммуномодулирующего действия пэгинтерферона, проявляющегося в предотвращении образования белков HBV и деплеции (истощении) внутрипеченочного пула кзкДНК [6] [7] [8] [9]. Тем не менее подход нуждается в дополнительных уточняющих исследованиях.
Помимо функционального излечения хронического вирусного гепатита B, существует куда менее достижимая цель стерильного излечения, когда HBsAg не обнаруживается, а ДНК HBV, включая кзкДНК и интегрированную, уничтожена.
В клиническом испытании B-Clear (NCT04449029) бепировирсен продемонстрировал высокую эффективность лечения, если отталкиваться от того факта, что потеря HBsAg установлена для почти трети пациентов после относительно короткого 24-недельного курса лечения.
Вирусологический ответ был зафиксирован среди как HBеAg-отрицательных пациентов, так и придерживающихся NA-терапии HBеAg-положительных. Это свидетельствует о том, что целевая для бепировирсена терапевтическая мРНК-последовательность HBV присутствует даже тогда, когда HBsAg получен из интегрированных вирусных геномов [10].
Отмеченный рост уровня АЛТ, сопутствовавший снижению уровня HBsAg, указывает на благотворные иммунные процессы. Известно, что повышение уровня АЛТ, суррогатного маркера воспаления печени, непрямым образом отражает факт иммуноопосредованного разрушения и клиренса инфицированных гепатоцитов и является предиктором потери HBsAg [11] [12].
Нельзя сказать, что нынешние результаты клинической проверки бепировирсена оказались разочаровывающими. Да, статус функционального излечения хронического вирусного гепатита B, подтвержденный отсутствием HBsAg и ДНК HBV на протяжении 6 месяцев после завершения лечения, зафиксирован у максимум 10% и 14% пациентов — соответственно среди проходивших фоновую терапию NA и без таковой. Однако с учетом короткого курса лечения эффективность следует воспринимать с должным оптимизмом.
Поскольку бепировирсен проявил наибольшую эффективность среди пациентов с изначально низким уровнем HBsAg, в дальнейшем следует рассматривать последний как основополагающий критерий выбора подходящих больных, которые с повышенной вероятностью извлекут пользу от лечения.
Кроме того, одним из предикторов успеха является статус HBeAg. В группе 1 среди HBeAg-отрицательных участников функционально излечились 10% и 14% — соответственно среди проходивших фоновую терапию NA и без таковой. При HBeAg-положительном статусе излечение отмечено для 6% и 0%.
К счастью, наблюдается тенденция, что большинство пациентов с хроническим вирусным гепатитом B являются HBeAg-отрицательными, то есть ДНК-последовательности HBV интегрированы в геном хозяина и являются основным источником HBsAg [10] [13].
Несмотря на множество изучаемых экспериментальных подходов к лечению хронического вирусного гепатита B [14], в их отношении назревает критика: мол, большинство из них фундаментально заблуждаются [15]. Инфекция HBV характеризуется очень высокой степенью генетической пластичности (тысячи квазивидов существуют у каждого отдельного пациента) [16], что является результатом отсутствия коррекционной активности обратной транскриптазы HBV, высокой скорости обновляемости (turnover) кзкДНК и постоянного иммунного давления, оказываемого на вирус. Учитывая, что даже одиночные точечные мутации HBV отменяют способность антисмысловых олигонуклеотидов (ASO) и малых интерферирующих РНК (миРНК) к специфическому расщеплению мРНК [17], под сомнение ставится даже теоретическая состоятельность данных классов лекарственных соединений для лечения хронического вирусного гепатита B.
На вышесказанное намекает тот факт, что снижение уровня HBsAg, обеспеченное бепировирсеном, оказалось сильнее среди пациентов с изначально более низким уровнем HBsAg (≤ 3000 МЕ/мл), что противоречит заявленному механизму действия препарата: сила снижения HBsAg не должна зависеть от его исходного уровня.
В предшествовавшем клиническом испытании NCT02981602 фазы IIa наблюдалась аналогичная картина [18]. Опять же, GSK3389404, вариант бепировирсена, конъюгированный с N-ацетилгалактозамином (GalNAc) в целях более таргетной доставки в гепатоциты [19], не оказал существенного влияния на снижение уровня HBsAg [20].
«Бизенгри» (Bizengri, зенокутузумаб) — первый лекарственный препарат, предназначенный для лечения онкологических заболеваний, опухоли которых характеризуются слияниями гена нейрегулина-1 (NRG1).
ОСНОВНЫЕ ФАКТЫ
В начале декабря 2024 года Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) одобрило «Бизенгри» в лечении взрослых пациентов с распространенным, неоперабельным или метастатическим немелкоклеточным раком легкого (НМРЛ) или распространенной, неоперабельной или метастатической аденокарциномой поджелудочной железы, если их опухоли несут NRG1-слияния и прогрессировали во время или после прохождения системной терапии [1].
Регуляторный вердикт вынесен в условном порядке, то есть препарату еще предстоит окончательно подтвердить свою эффективность.
Зенокутузумаб (zenocutuzumab), разработанный нидерландской «Мерус» (Merus), является биспецифическим моноклональным антителом против HER2 и HER3.
«Мерус» не намерена заниматься дальнейшим развитием зенокутузумаба: коммерческие права на лекарственное средство переданы «Патне терапьютикс» (Partner Therapeutics) [2].
В дальнейшем, не исключено, список показаний зенокутузумаба будет расширен за счет лечения других онкологических заболеваний с опухолевыми NRG1-слияниями.
ПОЧЕМУ ЭТО ВАЖНО
Таргетное лечение онкологических заболеваний постепенно расширяет свой фармакологический арсенал: когда лекарственному средству есть, за что «зацепиться», нацелившись на специфические онкогенные драйверные мутации, тогда и терапевтические исходы оказываются лучше, нежели в случае стандартной химиотерапии.
Так, в случае НМРЛ сейчас доступны препараты, направленные против активирующих геномных альтераций (мутаций, слияний, перестроек, транслокаций), которые поддерживают трансформацию, рост и прогрессирование опухоли и которые затрагивают гены EGFR, ALK, BRAF, ROS1, NTRK, RET, MET, KRAS или HER2.
При раке поджелудочной железы подобная таргетная терапия представлена лишь нацеливанием на мутации BRCA1/BRCA2.
КАК ЭТО РАБОТАЕТ
Белок нейрегулин-1 (NRG1) представляет собой лиганд, который, связываясь с рецептором 3 фактора эпидермального роста человека (HER3), промотирует гетеродимеризацию HER2/HER3 и онкогенез, что приводит к опухолевому росту [1] [2]. Высокая экспрессия NRG1, возникающая в результате аутокринной сигнализации или амплификации одноименного гена, ассоциирована с плохим прогнозом при некоторых видах рака и устойчивостью к стандартным методам лечения [3].
NRG1-слияния встречаются весьма редко: согласно оценкам «Мерус», при НМРЛ их можно обнаружить в 0,3–3% случаев, при раке поджелудочной железы — 0,5–1,5% случаев [4].
Зенокутузумаб (zenocutuzumab, MCLA-128) — биспецифическое моноклональное IgG1-антитело, которое связывается с внеклеточными доменами HER3 и рецептора 2 фактора эпидермального роста человека (HER2). Противораковая активность зенокутузумаба реализуется за счет блокирования связывания NRG1 с HER3 и димеризации HER2/HER3: происходит подавление пролиферации опухолевых клеток и их выживаемости, опосредованных онкогенным сигнальным путем PI3K–AKT–mTOR [5] [6].
Уничтожение опухолевых клеток зенокутузумабом осуществляется путем привлечения естественных киллеров (NK). Зенокутузумаб наделен усиленной антителозависимой клеточноопосредованной цитотоксичностью (ADCC).
КЛИНИЧЕСКИЕ ПОДРОБНОСТИ
Продолжающееся клиническое испытание eNRGy (NCT02912949) фазы II изучает эффективность и безопасность зенокутузумаба в лечении неоперабельных, метастатических или прогрессирующих солидных опухолей с NRG1-слияниями [1].
Проверка зенокутузумаба при NRG1-положительном немелкоклеточном раке легкого (n=64) установила следующие терапевтические исходы [2]:
частота общего ответа (ORR): 33% (95% ДИ [здесь и далее]: 22–46), включая 1,6% полных ответов (CR) и 31% частичных ответов (PR);
длительность ответа (DOR): медианных 7,4 месяца (4,0–16,6);
пропорция пациентов с DOR ≥ 6 месяцев: 43%.
Обновленные данные (n=79) свидетельствуют об улучшении клинической результативности зенокутузумаба [3]:
ORR: 37% (27–49), все ответы частичные;
DOR: медианных 14,9 месяца (7,4–20,4);
пропорция пациентов с DOR ≥ 6 месяцев: 81% (60–92);
пропорция пациентов с DOR ≥ 12 месяцев: 57% (34–75).
Клинические исходы при назначении зенокутузумаба при NRG1-положительной аденокарциноме поджелудочной железы (n=30) оказались следующими [2]:
Инструкция по медицинскому применению лекарственного препарата «Бизенгри» идет с «чернорамочным» предупреждением о риске эмбриофетальной токсичности: в ходе лечения необходимо придерживаться эффективной контрацепции.
«Атиа фармасьютикалс» (Atea Pharmaceuticals) разработала новую лекарственную комбинацию, которая позволяет навсегда вылечить хронический вирусный гепатит C (ВГС) за 8 недель.
ОСНОВНЫЕ ФАКТЫ
Сочетание бемнифосбувира (bemnifosbuvir) и рузасвира (ruzasvir) успешно прошло среднестадийную клиническую проверку.
Схема лечения предполагает ежедневный пероральный прием двух препаратов на протяжении 8 или 12 недель — соответственно без цирроза печени и с циррозом.
Бемнифосбувир с рузасвиром эффективно срабатывают при инфекции ВГС любого генотипа (включая трудноизлечимый генотип 3), а также при наличии компенсированного цирроза печени (стадия фиброза F4).
Комбинация характеризуется более чем приемлемым профилем безопасности и переносимости и отсутствием взаимодействия с другими лекарствами.
В 2025 году начнется регистрационная клиническая программа фазы III.
БОЛЬШИЕ ЧИСЛА
Несмотря на доступность противовирусных препаратов прямого действия (ПППД), представляющих собой пероральные лекарственные комбинации, хронической инфекцией вирусного гепатита C (ВГС) страдают приблизительно 50 млн человек во всём мире [1].
Ежегодно случается 1 млн новых заражений ВГС. Среди основных способов инфицирования: небезопасные инъекции, небезопасное медицинское обслуживания, переливание непроверенной крови, употребление инъекционных наркотиков, сексуальные контакты — все пути передачи обусловлены контактом с зараженной кровью.
В 2022 году хроническая инфекция ВГС повлекла за собой 242 тыс. смертельных исходов: главным образом ввиду цирроза печени и гепатоцеллюлярной карциномы (первичного рака печени).
КАК ЭТО РАБОТАЕТ
Бемнифосбувир (bemnifosbuvir, AT-527, RO7496998), будучи перорально биодоступной гемисульфатной солью AT-511, пролекарства аналога гуанозинмонофосфата, в организме в несколько этапов метаболизируется до активного 5’-трифосфата AT-9010, который действует как ингибитор РНК-полимеразы неструктурного белка 5B (NS5B), подавляющий вирусную репликацию [1] [2].
In vitro бемнифосбувир в 10 раз более активен чем софосбувир (sofosbuvir) против лабораторных штаммов и клинических изолятов вируса гепатита C генотипов 1–5. Бемнифосбувир остается полностью активным в случае мутационных замен, связанных с резистентностью к софосбувиру (S282T), при этом его эффективность всё еще в 58 раз выше, чем у последнего. Бемнифосбувир характеризуется низким риском лекарственных взаимодействий.
Рузасвир (ruzasvir, AT-038, MK-8408) — ингибитор неструктурного белка 5A (NS5A), открытый «Мерк и Ко» (Merck & Co.), которая в декабре 2021 года лицензировала его «Атиа» [3].
КЛИНИЧЕСКИЕ ПОДРОБНОСТИ
Клиническое исследование NCT05904470 фазы II проверило эффективность и безопасность ежедневно назначаемой пероральной комбинации из 550 мг бемнифосбувира и 180 мг рузасвира в лечении хронического вирусного гепатита C (ВГС) среди взрослых пациентов (n=275) без цирроза печени (стадия фиброза F0–F3) или с компенсированным циррозом (F4).
После 8 недель лечения к первичной конечной точке эффективности, установленной устойчивой вирусной супрессией, подтвержденной по прошествии 12 недель после терапии (SVR12), вышли 98% испытуемых (n=208/213), строго придерживавшихся режима приема препаратов [1].
Даже при учете тех больных, которые по каким-либо причинам пропускали очередную дозу лекарств (таковых было 17%), эффективность излечения всё равно оставалась высокой: 95% (n=242/256).
Среди комплаентных пациентов с ВГС генотипа 1–4 и без цирроза печени показатель SVR12 оказался справедливым для 99% человек (n=178/179), с циррозом — 88% (n=30/34).
Применение бемнифосбувира с рузасвиром характеризовалось приемлемой переносимостью. Нежелательные явления (НЯ) носили в целом легко-умеренную степень выраженности. Не зарегистрировано НЯ, которые либо повлекли за собой прекращение лечения, либо привели к изменению лабораторных показателей.
Дополнительные клинические данные будут представлены позже.
ЧТО ДАЛЬШЕ
«Атиа» планирует к запуску опорную клиническую проверку фазы III, которая будет представлена двумя испытания с группой активного сравнения и которая охватит большее число пациентов.
При отсутствии цирроза печени лечение будет осуществляться на протяжении 8 недель, при его наличии — 12 недель в целях повышения частоты излечения хронической инфекции.
Для удобства пациентов отдельные бемнифосбувир и рузасвир объединены в одну таблетку с фиксированной дозой.
ЧТО ЕЩЕ
В середине сентября 2024 года бемнифосбувир не справился с клиническим исследованием SUNRISE-3 (NCT05629962) фазы III среди амбулаторных высокорисковых пациентов с легко-умеренной коронавирусной инфекцией COVID-19: назначение препарата два раза в день на протяжении пяти дней поверх стандартного лечения не обеспечило статистически значимого снижения риска госпитализации или смерти по прошествии 29 дней [1].
БИЗНЕС
Период защиты интеллектуальной собственности «Атиа» заявлен вплоть до 2042 года — с потенциальной возможностью ее продления.
«Атиа», уже заплатившая «Мерк и Ко» аванс за лицензию на рузасвир, обязуется выдавать ей определенные суммы по мере прохождения этапов разработки, а также отчислять роялти от реализации готового препарата.
«Атиа» была основана в 2014 году Жаном-Пьером Соммадосси (Jean-Pierre Sommadossi) [1], который в свое время учредил «Айденикс фармасьютикалс» (Idenix Pharmaceuticals) и был соучредителем «Фармассет» (Pharmasset): первое предприятие в августе 2014 года купила «Мерк и Ко» за $3,85 млрд [2], вторую компанию в январе 2012 года приобрела «Гилеад сайенсиз» (Gilead Sciences) за $11,2 млрд [3].
В конце мая 2023 года «Атиа» отвергла предложение «Концентра байосайенсиз» (Concentra Biosciences), пожелавшей ее поглотить за $480 млн [4].
Терапия переменным электрическим полем подтвердила свою эффективность в первоочередном лечении неоперабельного рака поджелудочной железы.
ОСНОВНЫЕ ФАКТЫ
«Новокьюэ» (Novocure) осуществила успешную клиническую проверку полей для лечения опухолей (TTFields) в задаче терапии первой линии местнораспространенной протоковой аденокарциномы поджелудочной железы.
Добавление TTFields к стандартному набору из гемцитабина и наб-паклитаксела обеспечило продление общей выживаемости.
«Новокьюэ» собирается отправить в адрес регуляторов соответствующее регистрационное досье.
КАК ЭТО РАБОТАЕТ
Терапия переменным электрическим полем, также называемая полями для лечения опухолей (TTFields), обращается к низкоинтенсивным электрическим полям промежуточной частоты (100–500 кГц) в целях лечения раковых заболеваний.
Электрические поля TTFields не оказывают существенного влияния на здоровые клетки, поскольку они обладают иными свойствами (включая скорость деления, морфологию и электрические свойства), чем раковые клетки. Уникальный частотный диапазон позволяет TTFields проникать через клеточную мембрану, а низкая интенсивность — избегать деполяризации нервов или мышц либо значительного нагрева [1] [2].
Механизм действия TTFields обусловлен вмешательством в процесс деления клеток, что приводит к нарушению выравнивания диполей и индукции диэлектрофореза критически важных молекул и органелл во время митоза. Антимитотические эффекты отражаются гибелью опухолевых клеток [3] [4] [5].
TTFields изменяют организацию и динамику цитоскелета, нарушая подвижность и миграцию раковых клеток, которые необходимы для метастазирования [6].
Опосредованное TTFields разрушение клеток активирует иммунную систему и запускает противоопухолевый клеточный ответ. Электрические поля приводят к анеуплоидии, стрессу эндоплазматического ретикулума и низкому уровню внутриклеточного АТФ, стимулируя аутофагический ответ в раковых клетках. Отмечаются характерные признаки иммуногенной клеточной смерти, такие как высвобождение HMGB1, обнажение кальретикулина на поверхности клеток и опосредованное аутофагией высвобождение АТФ [5] [7].
TTFields нарушают механизм репарации повреждений ДНК путем снижения регуляции генов, важных для репликации ДНК и путей ответа на повреждение ДНК в раковых клетках [8] [9].
КЛИНИЧЕСКИЕ ПОДРОБНОСТИ
Клиническое исследование PANOVA-3 (NCT03377491) фазы III изучило подключение технологии TTFields к стандартной терапии гемцитабином и наб-паклитакселом взрослых пациентов (n=571) с прежде нелеченной местнораспространенной протоковой аденокарциномой поджелудочной железы.
По прошествии наблюдений на протяжении 18 месяцев экспериментальное лечение снизило риск смерти на 18%, если сравнивать с назначением только гемцитабина и наб-паклитаксела: отношение риска (hazard ratio, HR) 0,82 (p=0,039) [1].
Медиана общей выживаемости (OS) вышла к 16,2 месяца — против 14,2 месяца в группе контроля.
Частота выживаемости на протяжении 12 и 24 месяцев улучшилась на 13% и 33% соответственно.
Дополнительные клинические данные будут раскрыты позже.
ЧТО ЕЩЕ
Продолжается клиническое испытание PANOVA-4 (NCT06390059) фазы II, в котором технология TTFields проверяется совместно с атезолизумабом, гемцитабином и наб-паклитакселом в первоочередном лечении метастатического рака поджелудочной железы.
РАНЕЕ
Практический дебют технологии TTFields состоялся в середине апреля 2011 года, когда Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) одобрило устройство NovoTTF-100A (NovoTTF) для лечения мультиформной глиобластомы после ее рецидива вслед за химиотерапией [1].
В начале октября 2015 года устройство «Оптьюн» (Optune) [сейчас называется «Оптьюн Джио» (Optune Gio)] получило регуляторное разрешение для первоочередной терапии глиобластомы вместе с назначением темозоломида [2].
В конце мая 2019 года устройство NovoTTF-100L [сейчас называется «Оптьюн Луа» (Optune Lua) было дозволено для первоочередного лечения неоперабельной местнораспространенной или метастатической злокачественной плевральной мезотелиомы — в сочетании с пеметрекседом и платиносодержащей химиотерапией [3].
В середине октября 2024 года устройство «Оптьюн Луа» расширило спектр показаний, добавив лечение метастатического немелкоклеточного рака легкого (НМРЛ), прогрессировавшего во время или после платиносодержащей химиотерапии, — в комбинации с блокатором PD-(L)1 или доцетакселом [4].
ОДНАКО
Несмотря на регуляторное одобрение, эффективность технологии TTFields остается спорной в медицинском сообществе [1].
«Революшн медисинс» (Revolution Medicines) готовится изменить стандарт терапии второй линии распространенного или метастатического рака поджелудочной железы.
ОСНОВНЫЕ ФАКТЫ
Экспериментальный RMC-6236 — прямой ингибитор всех основных форм онкогенных белков семейства RAS, ключевого драйвера прогрессирования протоковой аденокарциномы поджелудочной железы.
Пероральный препарат-кандидат, наделенный приемлемой переносимостью, обеспечил существенное продление жизни в сравнении с любым ныне применяемым химиотерапевтическим режимом в условиях терапии второй линии.
Осталось дождаться завершения регистрационной клинической проверки, чтобы вывести новое лечение на рынок.
«Лечение рака поджелудочной железы десятилетиями остается одной из самых незакрытых потребностей медицины. Это самый RAS-мутантный из всех основных видов рака: опухоли с мутацией RAS встречаются у более чем 90% пациентов. Продемонстрированный уровень клинической активности RMC-6236 при его дозах с приемлемой переносимостью весьма примечателен».
Брайан Уолпин (Brian Wolpin), директор Центра по изучению рака желудочно-кишечного тракта (Gastrointestinal Cancer Center) и содиректор Центра по изучению опухолей поджелудочной железы и желчевыводящей системы (Pancreas and Biliary Tumor Center) при Институте рака Дана–Фарбер (Dana–Farber Cancer Institute, DFCI, Бостон, шт. Массачусетс, США), ведущий исследователь.
«Клинические данные RMC-6236 убедительно подтвердили показатели выживаемости без прогрессирования и общей выживаемости — однозначно важные для пациентов с раком поджелудочной железы, тем самым еще ближе приблизив нас к регуляторному утверждению препарата в качестве нового стандарта лечения при распространенном или метастатическом заболевании».
Марк Голдсмит (Mark Goldsmith), исполнительный директор и председатель правления «Революшн медисинс» (Revolution Medicines).
СУТЬ ВОПРОСА
Рак поджелудочной железы (РПЖ) — одно из самых смертоносных злокачественных новообразований, характеризующееся резистентность к стандартной химиотерапии и высокой летальностью. Пятилетняя выживаемость при отдаленном метастазировании составляет незначительных 3,1% [1].
Протоковая аденокарцинома поджелудочной железы (PDAC) и ее разновидности — наиболее распространенная форма рака поджелудочной железы: на ее долю выпадает 92% всех случаев последнего. По причине отсутствия ранних симптомов и методов выявления диагноз PDAC ставится на поздней или метастатической стадии 80% пациентов.
В более чем 90% случаев опухоли PDAC несут онкогенные мутации в семействе генов RAS, которые кодируют белки — малые ГТФазы. Самой распространенной при PDAC являются мутации KRASG12X (85%), включая KRASG12D (43%) [2].
КАК ЭТО РАБОТАЕТ
RMC-6236 — пероральный мощный прямой мультиселективный ингибитор RAS-сигнализации, подавляющий активность RAS-белков как дикого типа, так и мутантных вариантов канонических RAS-изоформ (HRAS, NRAS и KRAS). RMC-6236 проявляет противоопухолевую активность в том числе при таких онкогенных RAS-мутациях, как G12X, G13X и Q61X, распространенных при протоковой аденокарциноме поджелудочной железы, немелкоклеточном раке легкого, колоректальном раке [1] [2] [3].
КЛИНИЧЕСКИЕ ПОДРОБНОСТИ
Продолжающееся клиническое исследование NCT05379985 фазы I/Ib пригласило взрослых пациентов (n=127) с ранее леченой RAS-мутантной протоковой аденокарциномой поджелудочной железы.
Среди основных исходных характеристик испытуемых: медиана возраста 64 года (30–86), мужчин 56%, медианное число предшествовавших линий терапии 2 (1–11), медианное число предшествовавших курсов лечения по метастатическому показанию 0 (1% пациентов), 1 (45%), 2+ (54%), метастазы в печени (у 67%), метастатическое заболевание на стадии IV (у 52%). Генотипическое распределение RAS-мутаций: наиболее частыми были KRASG12X (у 84%), в том числе KRASG12D (32%), KRASG12V (32%) и KRASG12X (20%).
Монотерапия при помощи RMC-6236, назначаемого ежедневно перорально в дозе от 160 до 300 мг, обеспечила следующие исходы в ходе терапии второй линии [1]:
Медиана выживаемости без прогрессирования (PFS) вышла к 8,5 месяца (95% ДИ [здесь и далее]: 5,3–11,7) и 7,6 месяца (5,9–11,1) — соответственно в популяции пациентов с мутацией KRASG12X (n=42) и любой другой RAS-мутацией (n=57).
Медиана общей выживаемости (OS) составила 14,5 месяца (8,8–NE) и 14,5 месяца (8,8–NE), притом что частота 6-месячной OS получилась равной 89% (70–97) и 91% (77–96).
Частота общего ответа (ORR) достигла 29% и 22%, тогда как частота контроля заболевания (DCR) — 91% и 89%
Применение RMC-6236 характеризовалось приемлемой переносимостью. Среди наиболее распространенных нежелательных явлений (НЯ) в ответ на лечение: сыпь (у 98% пациентов), диарея (48%), тошнота (43%), рвота (31%), стоматит (31%), усталость (20%) — они носили главным образом легко-умеренную степень тяжести. Эти НЯ вынудили 35% участников снизить дозу RMC-6236 или временно прервать лечение.
ЧТО ДАЛЬШЕ
«Революшн» продолжает трехлетнее клиническое испытание RASolute 302 (NCT06625320) фазы III, в котором RMC-6236 (ежедневно по 300 мг) сравнивается со стандартной химиотерапией во второлинейном применении среди взрослых пациентов (n=460) с протоковой адернокарциномой поджелудочной железы, прошедшей один курс лечения по метастатическому показанию. Выбор химиотерапевтического режима отдан на выбор исследователя: гемцитабин + наб-паклитаксел (GnP), оксалиплатин + лейковорин + иринотекан + 5-фторурацил (mFOLFIRINOX), липосомальный иринотекан + 5-фторурацил + лейковорин (Nal-IRI+5-FU/LV) или оксалиплатин + лейковорин + 5-фторурацил (FOLFOX). Готовность результатов ожидается к середине 2026 года.
В долгосрочных планах стоит клиническая проверка RMC-6236 в первоочередном лечении протоковой адернокарциномы поджелудочной железы. Современные химиотерапевтические подходы при этом показании выдают медиану общей выживаемости в диапазоне 8,5–11,1 месяца [1] [2] [3].
ЧТО ЕЩЕ
«Революшн» тестирует RMC-9805, пероральный селективный ковалентный ингибитор мутантного белка KRASG12D, наиболее часто встречающегося при протоковой адернокарциноме поджелудочной железы. Намечено более тщательное клиническое изучение как монотерапии RMC-9805, так и сочетанного назначения вместе с RMC-6236; подобный коктейль призван обойти потенциальную лекарственную резистентность.
ЭКСПЕРТНЫЕ КОММЕНТАРИИ
Безо всяких сомнений противоопухолевая активность RMC-6236 получилась примечательной, если отталкиваться от клинических исходов лечения протоковой аденокарциномы поджелудочной железы при помощи стандартной химиотерапии, причем независимо от ее специфики.
Так, показатели ORR, PFS и OS, зарегистрированные в ходе клинических испытаний различных химиотерапевтических режимов терапии второй линии рака поджелудочной железы укладываются в пределы 3–17%, 2,0–3,5 месяца и 6,1–6,9 месяца [1] [2] [3] [4] [5] [6] [7]. Как видим, экспериментальный RMC-6236 с лихвой их превзошел, к примеру, обеспечив более чем двукратное продление жизни.
«Айкерво» (Iqirvo, элафибранор) — новый лекарственный препарат, предназначенный для лечения взрослых пациентов с первичным билиарным холангитом (ПБХ).
Первичный билиарный холангит (ПБХ) — редкое прогрессирующее аутоиммунное заболевание печени, ранее называвшееся первичным билиарным циррозом, характеризуется скоплением жёлчи и токсинов (холестаз) и хроническим воспалением, которые вызывают необратимый фиброз (рубцевание) печени и разрушение жёлчных протоков.
Элафибранор (elafibranor) — пероральный низкомолекулярный двойной агонист альфа- и дельта-рецепторов, активируемых пероксисомными пролифераторами (PPARα и PPARδ) [1].
«Айкерво» назначается в сочетании с урсодезоксихолевой кислотой (ursodeoxycholic acid, UDCA) при недостаточном терапевтическом ответе на нее или самостоятельно в случае непереносимости последней.
«Айкерво» одобрен Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) в начале июня 2024 года [2]. Регуляторный вердикт вынесен в условном порядке, то есть лекарственному средству предстоит подтвердить свою клиническую пользу, продемонстрировав, к примеру, продление жизни или предотвращение печеночной декомпенсации.
Элафибранор разработан французской «Дженфит» (Genfit), которая в середине декабря 2021 года передала французской «Ирсен» (Ipsen) права на коммерциализацию препарата за авансовых €120 млн, последующие выплаты до €360 млн по мере его реализации и двузначное роялти от продаж [3].
Для американских пациентов месячный курс лечения первичного билиарного холангита при помощи «Айкерво» обойдется в $11,5 тыс.
Согласно отраслевым прогнозам, пиковые продажи элафибранора достигнут $500 млн в год.
В клиническом исследовании ELATIVE (NCT04526665) фазы III (рандомизированном, двойном слепом, плацебо-контролируемом, многоцентровом, международном), охватившем взрослых пациентов (n=161) с ПБХ, ежедневное назначение 80 мг элафибранора на фоне UDCA на протяжении 52 недель обеспечило выход к биохимическому ответу для 51% испытуемых (n=55/108) — против 4% (n=2/53) в группе плацебо и UDCA (p<0,001) [4].
Под биохимическим ответом, как композитной конечной точкой, понималось сочетание следующих лабораторных показателей: снижение уровня сывороточной щелочной фосфатазы (СЩФ) ниже предела с максимумом в 1,67 раза выше верхней границы нормы (ВГН), не менее чем 15-процентное падение уровня СЩФ, снижение уровня общего билирубина хотя бы до ВГН.
Нормализация уровня СЩФ, как показателя прогрессирования поражения печени, начала отмечаться уже после 4 недель лечения и была зафиксирована для 15% пациентов (n=16/108) — против 0%(n=0/53) [p=0,002].
Применение элафибранора не привело к статистически значимому (p=0,20) ослаблению кожного зуда среди пациентов, страдающих умеренно-тяжелым таковым (балл ≥ 4): снижение на 1,93 балла по числовой рейтинговой шкале наихудшего зуда (WI-NRS) — против снижения на 1,15 балла.
Однако пациенты всё равно сообщили о заметном подъеме качества жизни, прежде нарушенном ввиду обременительности кожного зуда. Улучшения касались выраженности зуда, продолжительности и непрерывности сна, эмоционального воздействия зуда [5] [6].
Среди наиболее распространенных нежелательных явлений (НЯ) в ответ на назначение элафибранора: увеличение массы тела, абдоминальная боль, диарея, тошнота, рвота.
Продолжительные наблюдения установили, что по прошествии 78 недель лечения ПБХ элафибранором устойчивый биохимический ответ был зарегистрирован для 70% пациентов (n=19/27) против 0% (n=0/13), а нормализация СЩФ — для 19% (n=5/27) против 0% (n=0/13) [5] [7].
В середине августа 2024 года FDA одобрило «Ливделзи» (Livdelzi, селаделпар) — пероральный низкомолекулярный селективный агонист дельта-рецептора, активируемого пероксисомными пролифераторами (PPARδ). Селаделпар (seladelpar), разработанный «Симабей терапьютикс» (CymaBay Therapeutics), которую купила «Гилеад сайенсиз» (Gilead Sciences), является прямым конкурентом элафибранора.
«Ливделзи» (Livdelzi, селаделпар) — новый лекарственный препарат, предназначенный для лечения первичного билиарного холангита у взрослых.
ОСНОВНЫЕ ФАКТЫ
Первичный билиарный холангит (ПБХ; ранее назывался первичным билиарным циррозом) — редкое прогрессирующее аутоиммунное заболевание печени, приводящее к фиброзу, циррозу и преждевременной смерти.
Не существует способов, позволяющих окончательно вылечить ПБХ.
Нормализация биомаркеров холестаза вкупе с ослаблением выраженности зуда, обеспечиваемые селаделпаром, представляют собой явное изменение в парадигме лечения ПБХ.
Селаделпар назначается в сочетании с урсодезоксихолевой кислотой при недостаточном терапевтическом ответе на нее или самостоятельно в случае непереносимости последней.
Селаделпар не рекомендован к применению при декомпенсированном циррозе (асцит, варикозное кровотечение, печеночная энцефалопатия).
«Ливделзи» одобрен Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) в середине августа 2024 года [1].
Регуляторное решение вынесено в условном порядке: на базе суррогатного биомаркера, представленного снижением уровня щелочной фосфатазы. Препарату предстоит окончательно подтвердить клиническую пользу.
Селаделпар разработан «Симабей терапьютикс» (CymaBay Therapeutics), которую в конце марта 2024 года купила «Гилеад сайенсиз» (Gilead Sciences) за $4,3 млрд [2] [3].
Американским пациентам месячный курс лечения первичного билиарного холангита при помощи «Ливделзи» обойдется в $12,6 тыс., что на 10% и 30% дороже, чем «Айкерво» (Iqirvo, элафибранор) и «Окалива» (Ocaliva, обетихолевая кислота), другие препараты против ПБХ.
ПРЯМАЯ РЕЧЬ
«Диагноз первичного билиарного холангита ставится всё большему числу людей. Пациенты страдают от непрекращающегося зуда или ощущения мурашек по коже, а также изнурительной усталости, которая усугубляется зудом по ночам. Появление селаделпара — важная веха для всего сообщества».
Кэрол Робертс (Carol Roberts), президент PBCers Organization (США), некоммерческой организации поддержки пациентов с ПБХ.
«Селаделпар выводит лечение ПБХ на качественно новый уровень, поскольку существующие препараты, хронически принимаемые для замедления поражения печени и сдерживания прогрессирования, не помогают в 40% случаев. У многих пациентов сохраняются ненормальные печеночные показатели и не исчезает зуд, один из основных симптомов».
Палак Триведи (Palak Trivedi), ученый-клиницист и гепатолог из Бирмингемского университета (Великобритания).
«Селаделпар — несомненный признак медицинского прогресса, знаменующий наступление новой эры в лечении ПБХ, которое одновременно предоставляет положительные биохимические результаты и улучшает качество жизни, что является весьма желательными совпадением целей клиницистов и пациентов».
Дэвид Ассис (David Assis), гепатолог из Йельского медицинского института (Нью-Хейвен, шт. Коннектикут, США).
«Селаделпар — потенциально лучший в своем классе препарат, который изменяет сложившуюся картину течения ПБХ, не только улучшая и даже нормализуя показатели печеночной функции, но и облегчая кожный зуд как особенно тяжелый симптом».
Тимоти Уоткинс (Timothy Watkins), вице-президент по клинической разработке противовоспалительных лекарств «Гилеад сайенсиз» (Gilead Sciences).
«Люди, живущие с ПБХ, много лет ждали каких-либо изменений в стандартах лечения. Регуляторное одобрение многообещающего «„Ливделзи“ открыло новую страницу в медицинских подходах к ведению этой обременяющей болезни».
Дэниел О’Дэй (Daniel O’Day), председатель правления и исполнительный директор «Гилеад сайенсиз» (Gilead Sciences).
СУТЬ ВОПРОСА
Первичный билиарный холангит — редкое прогрессирующее аутоиммунное заболевание печени, при котором негнойный холангит (воспаление жёлчных протоков) по неизвестной причине поражает преимущественно междольковые жёлчные протоки, что приводит к портальной гипертензии и циррозу печени [1] [2].
Первичным билиарным холангитом страдают в основном женщины старше 40 лет [3]. Заболеваемость составляет 0,33–5,8 на 100 тыс. человеко-лет, распространенность — 1,91–40,2 на 100 тыс. человек [4].
Патогенез первичного билиарного холангита изучен не до конца и, по-видимому, включает генетическую предрасположенность и факторы окружающей среды [5] [6] [7].
Холестатическое поражение печени характеризуется различной скоростью иммуноопосредованного разрушения внутрипеченочных жёлчных протоков, сопровождающегося портальным воспалением [8] [9] [10] [11] [12]. Дуктопения (потеря жёлчных протоков) приводит к холестазу (нарушению оттока желчи из печени в двенадцатиперстную кишку) и гепатоцеллюлярному повреждению, прогрессирующему поражению печени с развитием фиброза, конечной стадии болезни печени, печеночной недостаточности [13] [12] [14] [15]. Независимо от стадии заболевания многие люди испытывают значительное ухудшение качества жизни, особенно из-за утомляемости и зуда [2] [8] [14].
Первичный билиарный холангит ассоциирован с другими аутоиммунными состояниями, такими как склеродермия, синдром Шегрена, саркоидоз, аутоиммунный гепатит.
В отличие от других причин цирроза, портальная гипертензия и варикозное расширение вен пищевода могут развиваться до появления цирроза [10] [11].
Гистологическое повреждение, характеризующееся гранулематозным лимфоцитарным холангитом, ассоциировано с ненормальными печеночными пробами, включая повышение уровня сывороточной щелочной фосфатазы (СЩФ), гамма-глутамилтрансферазы (ГГТ), активности аминотрансфераз и общего билирубина [16]. Эти биохимические показатели болезни коррелируют с тяжестью течения первичного билиарного холангита, эффективностью его лечения и исходами [17] [18] [19] [20] [21].
Уровень СЩФ как минимум в 1,67 раза выше верхней границы нормы (ВГН) и уровень общего билирубина выше ВГН служат суррогатными показателями активности заболевания, ассоциированы с риском прогрессирования болезни, с достаточной вероятностью предсказывают клиническую пользу лечения [18] [22]. Продемонстрировано, что исходы улучшаются при нормализации уровней СЩФ и общего билирубина [20].
Первоочередное лечение первичного билиарного холангита обращается к назначению урсодезоксихолевой кислоты с последующим, если биохимический ответ недостаточен, добавлением обетихолевой кислоты или фибратов (например, безафибрата, фенофибрата) [10] [11] [12] [23]. Терапии усталости не предусмотрено, а зуд лечится с переменным успехом [24]. Многим пациентам в конечном итоге может потребоваться трансплантация печени [25] [12] [14].
БОЛЬШИЕ ЧИСЛА
Согласно отраслевым прогнозам, селаделпар выйдет на уровень ежегодного заработка в $650 млн, а пиковых $2,9 млрд достигнет к 2036 году.
Оптимистичные предположения отталкиваются от допущения, что значительная часть пациентов, принимающих препарат «Окалива» (Ocaliva, обетихолевая кислота) авторства «Интерсепт фармасьютикалс» (Intercept Pharmaceuticals), которую в ноябре 2023 года купила итальянская «Альфасигма» (Alfasigma) за $800 млн [1] [2], перейдет на селаделпар или будет использовать его в качестве дополнительного лекарственного средства. Но это непростая задача, ведь обетихолевая кислота (obeticholic acid), агонист фарнезоидного X-рецептора (FXR), в течение ближайших восьми лет лишится патентной защиты, что породит ее недорогие генерические копии.
Тем не менее все предпосылки высокого интереса к селаделпару существуют. Во-первых, неоптимальный ответ на первоочередное назначение урсодезоксихолевой кислоты отмечается у 15–40% пациентов [3] [4], притом что 3–5% сталкиваются с неприемлемыми нежелательными явлениями [4]. Во-вторых, биохимический ответ на применение обетихолевой кислоты регистрируется у менее чем 50% больных [5].
КАК ЭТО РАБОТАЕТ
Селаделпар (seladelpar, MBX-8025) — мощный селективный агонист дельта-рецептора, активируемого пероксисомными пролифераторами (PPARδ), снижающий уровень биохимических маркеров холестаза, повреждения печени и ее воспаления [1] [2] [3].
PPARδ, активируемый жирными кислотами транскрипционный фактор, участвует в метаболизме жирных кислот и воспалении [4] [5] [6]. В печени гены, регулируемые PPARδ, экспрессируются в гепатоцитах, холангиоцитах, клетках Купфера и звёздчатых клетках [7] [8]. PPARδ играет критическую роль в гомеостазе жёлчных кислот и оказывает противофиброзное действие [9] [4] [5] [6] [10].
Агонизм PPARδ ассоциирован со снижением синтеза жёлчных кислот, подавлением воспалительных цитокинов, ингибированием пролиферации и активации звёздчатых клеток.
Оригинатором селаделпара является «Орто-МакНил-Янссен фармасьютикалс» (Ortho-McNeil-Janssen Pharmaceuticals), подразделение «Джонсон энд Джонсон» (Johnson & Johnson), которое в августе 2006 года лицензировало молекулу «Симабей терапьютикс» (CymaBay Therapeutics), на тот момент называвшейся «Метаболекс» (Metabolex) [11].
КЛИНИЧЕСКИЕ ПОДРОБНОСТИ
Клинические исследования ENHANCE (NCT03602560) и RESPONSE (NCT04620733) фазы III (рандомизированные, двойные слепые, плацебо-контролируемые, многоцентровые, международные) охватили взрослых пациентов (n=265 и n=193) с первичным билиарным холангитом и недостаточным ответом на назначение урсодезоксихолевой кислоты или с ее непереносимостью.
Среди основных требований к участникам: уровень сывороточной щелочной фосфатазы (СЩФ) как минимум в 1,67 раза выше верхней границы нормы, несмотря на назначение урсодезоксихолевой кислоты на протяжении хотя бы 12 месяцев.
Испытуемые получали ежедневно перорально плацебо или селаделпар в дозе 5 мг (только в ENHANCE) или 10 мг — на протяжении 3 или 12 месяцев в ENHANCE и RESPONSE соответственно.
Композитная первичная конечная точка эффективности лечения ПБХ была установлена пропорцией пациентов, биохимически ответивших на терапию. Ответ фиксировался при удовлетворении всем следующим критериям: снижение уровня СЩФ ниже предела с максимумом в 1,67 раза выше верхней границы нормы (ВГН), не менее чем 15-процентное падение уровня СЩФ, снижение уровня общего билирубина хотя бы до ВГН.
К указанному показателю эффективности вышли 78% (n=43/55) и 62% (n=79/128) пациентов в группах, получавших 10 мг селаделпара, — против 13% (n=7/56) и 20% (n=13/65) в группах плацебо (p<0,0001) [1] [2] [3].
Антихолестатический эффект селаделпара подтвердился нормализацией уровня СЩФ (не выше верхней границы нормы) у 27% (n=15/55) и 25% больных (n=32/128) — против 0% (n=0/56 и n=0/65) в контрольных группах (p<0,0001) [2].
Снижение уровня СЩФ составило 44% и 42% — против 4% (p<0,0001).
Применение селаделпара привело к значительному улучшению сывороточных биомаркеров повреждения печени и липидного профиля, таких как АЛТ, ГГТ, холестерин ЛПНП, триглицериды.
Назначение селаделпара обеспечило ослабление зуда: среди пациентов с зудом умеренно-тяжелой степени выраженности (балл ≥ 4 по числовой рейтинговой шкале, NRS) его снижение составило 3,1 и 3,2 пункта (после 3 и 6 месяцев лечения в ENHANCE и RESPONSE) — против снижения на 1,6 и 1,7 пункта в группах плацебо (p=0,02 и p<0,005).
Профиль безопасности селаделпара не характеризовался какими-либо серьезными нежелательными явлениями (НЯ), на которые следовало бы обратить особое внимание.
КОНТРАРГУМЕНТЫ
Селаделпару придется напрямую соперничать с «Айкерво» (Iqirvo), элафибранор), в начале августа 2024 года одобренным FDA с подачи французских «Дженфит» (Genfit) и «Ирсен» (Ipsen) при таком же показании для лечения первичного билиарного холангита.
Если сравнивать терапевтическую эффективность селаделпара и элафибранора (elafibranor), двойного агониста PPARα и PPARδ, хотя методологически это не совсем верно, получается, что выход к композитному биохимическому ответу с коррекцией на плацебо был справедлив для соответственно 42% (95% ДИ [здесь и далее]: 28–53) и 47% (32–57) пациентов, а нормализация СЩФ — для 25% (18–33) и 15% (6–23). Выраженность зуда снизилась на абсолютных 3,2 и 1,9 балла.
Зуд является частым симптомом у пациентов с ПБХ и связан с высокой концентрацией в плазме крови веществ, которые выводятся с желчью. При гепатоцеллюлярной недостаточности зуд, как правило, стихает, что позволяет предположить, что холестатическая печень ответственна за выработку пруритогенных веществ [1].
Элафибранор для лечения ПБХ, когда урсодезоксихолевая кислота не справляется.
Почему назначение селаделпара привело к статистически значимому уменьшению зуда, тогда как применение элафибранора этого не обеспечило?
В исследовании селаделпара у 14% пациентов был цирроз печени, а исходная усредненная интенсивность зуда (среди участников с умеренно-тяжелым таковым) составляла 6,1±1,4 балла в группе препарата и 6,6±1,4 балла в группе плацебо, согласно числовой рейтинговой шкале (NRS). В исследовании элафибранора у 43% испытуемых был мостовидный фиброз или цирроз, а изначальная усредненная интенсивность зуда составляла соответственно 3,3±2,8 и 3,2±2,9 балла, согласно числовой рейтинговой шкале наихудшего зуда (WI-NRS). Помимо различий в дизайне исследований уместно задаться вопросом: связано ли отсутствие преимущества в ослаблении зуда при использовании элафибранора с механистическими особенностями препарата или всё же с более высоким процентом пациентов с циррозом?
Что касается внедрения селаделпара и элафибранора в клиническую практику, в дальнейшем уместно рассмотреть возможность их использования в качестве препаратов первой линии, то есть вместо урсодезоксихолевой кислоты, потому что до 40% пациентов к ней рефрактерны [2], а выдаваемая ею частота биохимического ответа (значительное снижение уровня СЩФ на фоне нормализации уровня общего билирубина), который ассоциирован с хорошим долгосрочным прогнозом [3], не сильно отличается от таковой при применении этих новых препаратов.
Не исключено, разумным окажется первоочередное назначение комбинации из урсодезоксихолевой кислоты и селаделпара или элафибранора.
Селаделпар и элафибранор — новые и запатентованные лекарственные соединения, и потому дорогостоящие в приобретении и присутствующие не на любом рынке. Приемлемым решением является добавление фибратов (давно ставших генерическими, и потому доступных по цене) к урсодезоксихолевой кислоте [4] [5]. Это подтверждено рядом исследований и метаанализов, изучивших подобное применение фибратов вне инструкции: агониста PPARα фенофибрата (fenofibrate) [6] [7] [8] [9] и пан-агониста PPAR безафибрата (bezafibrate) [10] [11] [12] [13] [14]. Добавление фибратов также поможет снять зуд [15].
ЧТО ДАЛЬШЕ
Селаделпар изучается в ходе долгосрочной проверки безопасности и эффективности лечения первичного билиарного холангита в рамках пятилетнего клинического испытания ASSURE (NCT03301506) фазы III среди пациентов из других исследований, пожелавших продолжить терапию.
Согласно промежуточному анализу, охватившему данные за 2 года применения селаделпара, 70% участников (n=99/179) вышли к вышеописанной композитной конечной точке эффективности лечения. При этом у 42% человек отмечена нормализация СЩФ [1].
Организовано трехлетнее клиническое испытание AFFIRM (NCT06051617) фазы III среди пациентов с ПБХ и компенсированным циррозом. Проверяется гипотеза, способен ли селаделпар отсрочить время до наступления одного из следующих событий: смерть по любой причине, трансплантация печени, балл по шкале MELD (модель для оценки терминальной стадии заболевания печени) ≥ 15, требующий лечения асцит, госпитализация по причине печеночной декомпенсации (кровотечение в местах варикозного расширения вен пищевода или желудка, печеночная энцефалопатия, спонтанный бактериальный перитонит).
Результаты AFFIRM, которые должны быть представлены в FDA до февраля 2030 года, лягут в основу полноценного одобрения «Ливделзи».
Клиническая проверка показала, что сочетание препаратов «Опдиво» (Opdivo, ниволумаб) и «Ервой» (Yervoy, ипилимумаб), назначаемое в ходе первоочередного лечения неоперабельного рака печени (гепатоцеллюлярной карциномы), превосходит эффективность, обеспечиваемую терапией в лице препарата «Ленвима» (Lenvima, ленватиниб) или «Нексавар» (Nexavar, сорафениб).
ОСНОВНЫЕ ФАКТЫ
«Бристол-Майерс Сквибб» (Bristol-Myers Squibb) продемонстрировала преимущество сочетания из ниволумаба (nivolumab) и ипилимумаба (ipilimumab), блокаторов PD-1 и CTLA-4, над ленватинибом (lenvatinib) или сорафенибом (sorafenib), тирозинкиназными ингибиторами, продвигаемыми соответственно «Эйсай» (Eisai) / «Мерк и Ко» (Merck & Co.) и «Байер» (Bayer), в ходе перволинейной терапии рака печени.
По отношению к препаратам сравнения иммунноонкологический коктейль снизил риск смерти на 21% и снизил риск прогрессирования заболевания или смерти на 13%.
Этого недостаточно, чтобы опередить нынешний стандарт в лице комбинации из «Тецентрика» (Tecentriq, атезолизумаб) и «Авастина» (Avastin, бевацизумаб) — блокатора PD-L1 и ингибитора VEGF авторства «Рош» (Roche).
Однако в абсолютном исчислении продление общей выживаемости оказалось превосходным.
Регистрационное досье отправлено в адрес регуляторов.
ПРЯМАЯ РЕЧЬ
«Медиана общей выживаемости получилась одной из самых длинных, которые мы когда-либо наблюдали в ходе лечения распространенной гепатоцеллюлярной карциномы. Мы уверены, что разработали новый стандарт лечения».
Питер Галле (Peter Galle), клинический гепатолог из Медицинского центра при Майнцском университете (земля Рейнланд-Пфальц, Германия).
«Отмеченная нами частота уменьшения опухоли — одна из самых высоких среди других вариантов лечения рака печени. Высокий уровень ответа на терапию повышает шансы трансформации заболевания из неоперабельного в поддающееся резекции».
Лаура Гофф (Laura Goff), исполнительный медицинский директор Центра ухода за онкологическими пациентами при Онкологическом центре Вандербильта — Инграма (VICC, шт. Теннесси, США).
«Несмотря на продолжающееся развитие фармакологической науки, прогноз для пациентов с гепатоцеллюлярной карциномой по-прежнему остается плохим. Вот почему важно предложить им новые способы лечения, которые, возможно, помогут».
Дана Уолкер (Dana Walker), вице-президент и руководитель глобальной программы по раку желудочно-кишечного тракта и мочеполовой системы «Бристол-Майерс Сквибб» (Bristol-Myers Squibb).
КЛИНИЧЕСКИЕ ПОДРОБНОСТИ
Клиническое исследование CheckMate 9DW (NCT04039607) фазы III (рандомизированное, открытое, с активным контролем, многоцентровое, международное) пригласило взрослых пациентов (n=668) с распространенной гепатоцеллюлярной карциномой, ранее не проходившей системной терапии.
Участникам назначали либо комбинацию из ниволумаба и ипилимумаба, либо ленватиниб или сорафениб (на выбор исследователя) — до момента прогрессирования заболевания или неприемлемой токсичности.
После наблюдений на протяжении медианных 35,2 месяца (26,8–48,9) результаты получились следующими [1] [2] [3].
Общая выживаемость в группе «Опдиво» с «Ервоем» вышла к 23,7 месяца (95% ДИ [здесь и далее]: 18,8–29,4) — против 20,6 месяца (17,5–22,5) в группе «Ленвимы» или «Нексавара». Риск смерти снизился на относительный 21%: отношение риска (hazard ratio, HR) 0,79 (0,65–0,96); p=0,018.
Вероятность остаться в живых на протяжении 24 месяцев составила 49% против 39%, 36 месяцев — 38% против 24%.
Частота общего ответа (ORR) составила 36% (31–42), включая 7% полных ответов (CR), — против 13% (10–17), в том числе 2% CR (p<0,0001).
Медиана длительности ответа (DoR) получилась равной 30,4 месяца (21,2–NE) — против 12,9 месяца (10,2–31,2).
Медиана выживаемости без прогрессирования (PFS) обозначилась на уровне 9,1 месяца (6,6–10,5) — против 9,2 месяца (7,9–11,1). Риск прогрессирования заболевания или смерти снизился на относительных 13%: HR 0,87 (0,72–1,06). Статус PFS в течение 18 месяцев оказался справедливым для 34% пациентов против 18%, 24 месяцев — 28% против 12%.
Назначение иммуноонкологического коктейля привело к снижению риска ухудшения симптомов заболевания на относительных 24%: HR 0,76 (0,62–0,93); p=0,0059.
КОНТРАРГУМЕНТЫ
Использование ленватиниба или сорафениба в качестве контрольной группы — сомнительный выбор «Бристол-Майерс Сквибб». Разумнее было остановиться на более эффективных схемах, представленных либо сочетанием атезолизумаба (atezolizumab) с бевацизумабом (bevacizumab), за которым стоит «Рош», либо дуэтом «Имфинзи» (Imfinzi, дурвалумаб) с «Имджудо» (Imjudo, тремелимумаб) — блокатора PD-L1 с блокатором CTLA-4, продвигаемым «АстраЗенека» (AstraZeneca). Первая схема является предпочтительной, вторая выступает альтернативной в случае противопоказаний к назначению бевацизумаба или его непереносимости.
Согласно нынешним рекомендациям Американского общества клинической онкологии (ASCO), атезолизумаб с бевацизумабом лидирует по эффективности первоочередного лечения гепатоцеллюлярной карциномы, улучшая выживаемость и сдерживая прогрессирование заболевания относительно сорафениба: OS HR 0,66 (0,52–0,85) и PFS HR 0,65 (0,53–0,81) [1] [2].
Применение дурвалумаба (durvalumab) с тремелимумабом (tremelimumab) характеризуется только улучшением выживаемости относительно сорафениба: OS HR 0,78 (0,67–0,92) и PFS HR 0,90 (0,77–1,05) [3] [4].
ЭКСПЕРТНЫЕ КОММЕНТАРИИ
Начиная с марта 2020 года, «Опдиво» с «Ервоем» применяются во второлинейной терапии гепатоцеллюлярной карциномы. Но бизнес «Бристол-Майерс Сквибб» требует расширения охвата пригодных пациентов.
Результаты клинической проверки этого сочетания в перволинейной терапии рака печени получились на первый взгляд идентичными таковым в случае перволинейного использования дурвалумаба с тремелимумабом. Но есть важные отличия.
Во-первых, «Опдиво» с «Ервоеем» обеспечили более продолжительную общую выживаемость с медианой 20,6 месяца — против 16,4 месяца в случае «Имфинзи» с «Имджудо».
Во-вторых, они повысили шансы остаться в живых: до 49% и 38% на протяжении 24 и 36 месяцев — против 41% и 31%.
В-третьих, на лечение ответила большая пропорция пациентов: ORR 36% и CR 7% — против ORR 20% и CR 3%.
В-четвертых, ответ на лечение продолжался дольше: 30,4 месяца — против 22,3 месяца.
Однако справиться с прогрессированием заболевания удалось аналогично плохо.
Осталось разобраться с нежелательными явлениями и их частотой, чтобы понять, насколько новая схема лечения окажется переносимой.
Перспективность ниволумаба с ипилимумабом в контексте первоочередного лечения гепатоцеллюлярной карциномы сомнений не вызывает, ведь долгосрочные 4-летние наблюдения за пациентами, получавшими дурвалумаб с тремелимумабом, установили прилично высокую пропорцию долгожителей, то есть перешагнувших отметку 36-месячной выживаемости: каждый четвертый (25%) — против 15% в группе сорафениба [1].
В идеале было бы правильным напрямую сравнить эти две схемы с «Тецентриком» и «Авастином», но гранды фармотрасли на такое вряд ли пойдут.
ОДНАКО
Списывать сорафениб или ленватиниб со счетов пока рано. Согласно систематическому обзору и метаанализу, сочетание тирозинкиназного ингибитора, блокатора PD-1 и локорегионарной терапии характеризовалось максимальными шансами на конверсию изначально неоперабельной гепатоцеллюлярной карциномы в поддающуюся радикальной резекции, то есть с потенциалом очень длительной выживаемости [1].
ЧТО ПРОИЗОШЛО. «Тевимбра» (Tevimbra, тислелизумаб) — новый лекарственный препарат, предназначенный для лечения неоперабельной или метастатической плоскоклеточной карциномы пищевода у взрослых после предшествовавшей системной химиотерапии, которая проводилась без блокатора PD-(L)1.
«Тевимбра», разработанный китайской «Бейджин» (BeiGene), одобрен Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) в середине марта 2024 года [1].
Европейское агентство по лекарственным средствам (EMA) выдало «Тевимбра» аналогичное маркетинговое разрешение чуть раньше — в середине сентября 2023 года [2].
ПОЧЕМУ ЭТО ВАЖНО. Рак пищевода является восьмым по распространенности онкологическим заболеванием в мире и шестой причиной смерти от онкопатологий [1]. Плоскоклеточная карцинома пищевода — самый распространенный подтип рака пищевода: на его долю приходится 85% случаев [2] [3]. Согласно прогнозам, в 2040 году число новых случаев рака пищевода составит 957 тыс., что на 60% больше, чем было зарегистрировано в 2020 году, свидетельствуя о необходимости дополнительных методов лечения [1]. На момент постановки диагноза рака пищевода у свыше двух третей пациентов он имеет прогрессирующий или метастатический характер. Ожидаемая пятилетняя выживаемость при раке пищевода с отдаленными метастазами не превышает 6% [4].
ЧТО ВЫЯСНИЛОСЬ. Клиническое исследование RATIONALE 302 (NCT03430843) фазы III (рандомизированное, открытое, с группой активного сравнения, многоцентровое, международное) охватило взрослых пациентов (n=512) с неоперабельной, местнораспространенной или метастатической плоскоклеточной карциномой пищевода, которая прогрессировала в ходе первоочередного лечения или после него.
Назначение тислелизумаба привело к улучшению исходов по сравнению с применением химиотерапии (паклитаксела, доцетаксела или иринотекана) [1]:
Медиана общей выживаемости (OS): 8,6 месяца — против 6,3 месяца. Риск смерти снизился на относительных 30%: отношение риска (hazard ratio, HR) 0,70 (95% ДИ [здесь и далее]: 0,57–0,85; p=0,0001).
Вероятность остаться в живых на протяжении 12 месяцев: 37% — против 24%.
Частота общего ответа (ORR): 20%, включая 2% полных ответов (CR) и 18% частичных ответов (PR), — против 9,8%, включая 0,4% CR и 9,4% PR.
Медиана длительности ответа (DoR): 7,1 месяца — против 4,0 месяца.
У пациентов с показателем PD-L1-позитивности опухолевой площади (TAP) ≥ 10% медиана OS составила 10,3 месяца — против 6,8 месяца: HR 0,54 (0,36–0,79; p=0,0006).
С нежелательными явлениями (НЯ), вызванными лечением и протекавшими в тяжелой форме, столкнулись 19% пациентов в группе тислелизумаба — против 56% в группе химиотерапии.
КАК ЭТО РАБОТАЕТ. Тислелизумаб (tislelizumab, BGB-A317) — гуманизированное моноклональное IgG4-антитело, которое связывает PD-1 на T-клетках, тем самым препятствуя его взаимодействию со своими лигандами, PD-L1 и PD-L2. В итоге разблокируются противоопухолевый иммунный ответ, пролиферация T-клеток и выработка цитокинов. Некоторые опухоли характеризуются повышенной регуляцией лигандов PD-1, что способствует подавлению активного T-клеточного иммунного надзора [1].
Тислелизумаб спроектирован так, чтобы минимально связывать Fc-гамма-рецептор 1 (FcγRI) на макрофагах — в целях ограничения антителозависимого клеточного фагоцитоза (ADCP) T-клеток макрофагами как потенциального механизма резистентности к анти-PD-1-терапии [2] [3] [4].
Тислелизумаб обладает более высоким сродством к PD-1, чем «Китруда» (Keytruda, пембролизумаб) и «Опдиво» (Opdivo, ниволумаб), блокаторы PD-1 авторства «Мерк и Ко» (Merck & Co.) и «Бристол-Майерс Сквибб» (Bristol-Myers Squibb), что может быть частично объяснено различными механизмами связывания с PD-1 [5] [6] [7].
ЧТО ДАЛЬШЕ. «Тевимбра» готовится расширить список показаний, подключив первоочередное лечение неоперабельной, рецидивирующей, местнораспространенной или метастатической плоскоклеточной карциномы пищевода и местнораспространенной неоперабельной или метастатической аденокарциномы желудка или пищеводно-желудочного соустья. К июлю и декабрю 2024 года FDA должно вынести соответствующие вердикты [1] [2].
EMA собирается выдать маркетинговое разрешение «Тизвени» (Tizveni, тислелизумаб) для лечения местнораспространенного или метастатического немелкоклеточного рака легкого (НМРЛ) по трем показаниям: в рамках первочередной терапии неплоскоклеточного и плоскоклеточного НМРЛ в сочетании с химиопрепаратами и в ходе монотерапии второй линии после провала платиносодержащей химиотерапии [3].
ТЕМ ВРЕМЕНЕМ. Дебют тислелизумаба в родном Китае состоялся в конце декабря 2019 года: «Байдзеань» (Baizean) предназначен для лечения рецидивирующей или рефрактерной классической лимфомы Ходжкина после провала как минимум двух линий химиотерапии [1] [2]. Впоследствии тислелизумаб прилично расширил список показаний, выступая своего рода местным аналогом «Китруды», который сейчас охватывает 35 показаний (17 типов опухолей и 2 показания вне привязки к анатомической локализации опухоли) и не собирается на этом останавливаться [3] [4]. На конец 2023 года тислелизумаб был одобрен для применения при 12 показаниях, в 2024 году ожидается добавление еще трех [5].
ОБРАТНАЯ СТОРОНА. Идея фикс тислелизумаба, который целиком и полностью придуман в Китае, состоит в том, чтобы избавить эту коммунистическую страну от кабалы дорогостоящих западных блокаторов PD-(L)1 [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11] [12] [13] [14]. Рано или поздно, но это обязательно случится, тем паче эффективность тислелизумаба не уступает американским лекарствам [15] [16] [17] [18] [19] [20] [21] [22] [23] [24] [25] [26] [27] [28] [29].
Китай буквально в приказном порядке вынуждает иностранных и местных фармпроизводителей резко сбрасывать ценники на лекарства, чтобы они имели хождение и страховое медицинское покрытие на столь лакомом ввиду своей масштабности рынке сбыта. И, что примечательно, на правительством уровне поощряет бурное развитие биотехнологий для разработки оригинальных препаратов, а не дженериков. Нет никак сомнений в серьезности, и главное, оправданности намерений: одних только блокаторов PD-(L)1 появилось 15 наименований за минувшие шесть лет, при этом самый дешевый стоит в 16 раз меньше, чем «Китруда» [30].
Могло бы показаться, что появление тислелизумаба на высокоприбыльных Западных рынках наделит «Бейджин» внушительным заработком. Но это не так, ведь в тех же США уже доступны девять блокаторов PD-(L)1. Китайскому фармпредприятию было бы правильным либо придерживаться стратегии сниженных цен, либо выбирать те показания, для которых препараты этого класса еще не одобрены. К примеру, «Кохерус байосайенсиз» (Coherus BioSciences) и «Шанхай Цзюньши байосайенсиз» (Shanghai Junshi Biosciences) выставили стоимость PD-1-блокатора «Локторзи» (Loqtorzi, торипалимаб) на 20% ниже, чем цена «Китруды» [31], и сориентировали его на лечение карциномы носоглотки, для которой прежде не было предложено иммунотерапии [32].