РЕЗЮМЕ
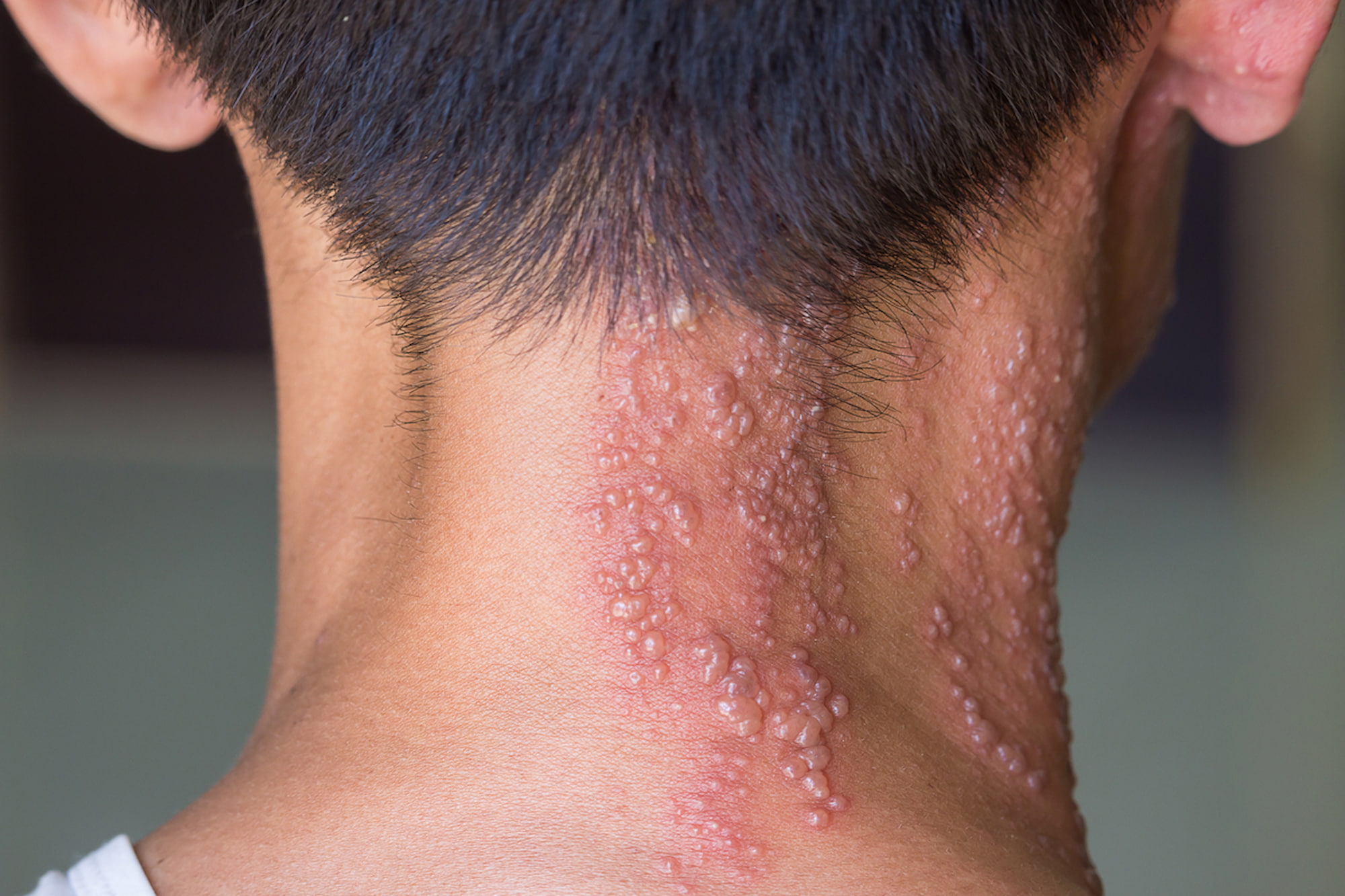
- Опоясывающий лишай (опоясывающий герпес) — распространенное заболевание, вызывающее сыпь и болевой синдром.
- С возрастом риск столкнуться с опоясывающим лишаем резко возрастает.
- «Шингрикс» (Shingrix, RZV) авторства «ГлаксоСмитКляйн» (GlaxoSmithKline) — единственная вакцина, которая предоставляет безоговорочно мощную и надежную защиту от опоясывающего лишая.
- После вакцинации «Шингрикс» иммунитет против опоясывающего лишая сохраняется на протяжении 10 лет и даже дольше.
- Фармотрасль продолжает искать варианты улучшить защитные показатели «Шингрикс».
ЧТО ПРОИЗОШЛО
Иммунная защита от опоясывающего лишая, активированная вакциной «Шингрикс» (Shingrix, RZV), сохраняется на протяжении не менее чем десяти лет после прививки.
«Шингрикс» — вакцина, предназначенная для профилактики опоясывающего герпеса и постгерпетической невралгии.
Сила превентивной защиты, предоставляемой «Шингрикс», близка к 90% (или даже превышает) и держится многие годы, причем вне зависимости от возраста (справедливо для лиц без ослабленного иммунитета).
Инактивированная рекомбинантная вакцина «Шингрикс», разработанная «ГлаксоСмитКляйн» (GlaxoSmithKline), вводится внутримышечно дважды — с интервалом от двух до шести месяцев.
До появления «Шингрикс» профилактика опоясывающего лишая была возможна только одной вакциной: живой аттенуированной «Зоставакс» (Zostavax, ZVL) авторства «Мерк и Ко» (Merck & Co.). Иммунизационная эффективность «Зоставакс» посредственна: она снижается риск развития заболевания на 51%, а постгерпетической невралгии на 67%.

ПОЧЕМУ ЭТО ВАЖНО
У переболевших (обычно в детстве) ветряной оспой (ветрянкой) вызвавший ее вирус остается персистировать в нейронах спинномозговых узлов и черепных нервов. В случае реактивации латентного (спящего) вируса ветряной оспы (герпесвирус человека типа 3, Human alphaherpesvirus 3; варицелла-зостер, Varicella zoster virus, VZV) возникает другая форма этой инфекции — опоясывающий лишай (опоясывающий герпес).
Вероятность развития опоясывающего лишая возрастает по мере снижения Т-клеточного иммунитета, поэтому в группе риска находятся люди пожилого возраста или с ослабленной иммунной системой [1], а также те, кто страдает хроническими заболеваниями, такими как хроническая обструктивная болезнь легких (ХОБЛ), сахарный диабет или астма [2].
Заболевание, так или иначе поражающее каждого третьего человека [3] [4] [5] [6], приводит к болевому синдрому (жжение, покалывание, онемение, пульсация, повышенная чувствительность, пульсация) и появлению волдырей на коже с дерматомным типом распределения [7] [8] [9] [10].
Сыпь при опоясывающем лишае обычно проходит через две–четыре недели, тогда как обременяющая постгерпетическая невралгия у одной пятой заболевших может сохраняться месяцами и годами после заживления кожных поражений.
Среди множества других осложнений: офтальмологический опоясывающий герпес, синдром Рамсея Ханта (ганглионит узла коленца лицевого нерва), миелопатия, асептический менингит, энцефалит, полинейропатия (включая синдром Гийена — Барре), полирадикулит, васкулопатия, острый некроз сетчатки, прогрессирующий некроз наружных слоев сетчатки.
Лечение опоясывающего лишая, которое следует начинать не позднее 72 часов после манифестации заболевания, предполагает применение системных противовирусных препаратов, таких как валацикловир (valaciclovir), фамцикловир (famciclovir), ацикловир (aciclovir), бривудин (brivudine), фоскарнет (foscarnet), аменамевир (amenamevir).
КАК ЭТО РАБОТАЕТ
«Шингрикс» (Shingrix, GSK1437173A, Hz/su) — инактивированная рекомбинантная субъединичная вакцина, сочетающая антиген в виде оболочечного гликопротеина E (gE) вируса ветряной оспы и адъювантную систему AS01B, улучшающую иммунологический ответ [1] [2] [3].
Иммунное воздействие белка gE стимулирует выработку антител против него, тем самым формируя адаптивный иммунитет против вируса.
Фирменный адъювант AS01B, заключенный в липосомальную рецептуру, комбинирует 3-О-дезацилированный 4′-монофосфорил-липид A (MPL) сальмонеллы (Salmonella minnesota) и молекулу сапонина (QS-21) из растительного экстракта квиллайи мыльной (Quillaja saponaria).
MPL, работающий как агонист толл-подобного рецептора 4 (TLR), стимулирует антигенпрезентирующие клетки посредством активации врожденного иммунитета. QS-21 действует как стимулятор путей врожденного иммунитета в моноцитах. Липосомальная формуляция нивелирует присущую QS-21 гемолитическую активность, а также усиливает презентацию антигена по сравнению с эмульсионной рецептурой AS02 [4] [5] [6] [7] [8] [9].
ДОСТУПНОСТЬ
Коммерческий дебют вакцины «Шингрикс» (Shingrix, RZV) состоялся в Канаде в середине октября 2017 года [1].
В США «Шингрикс» получила одобрение Управления по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) дважды. Вначале, в конце октября 2017 года, для профилактики опоясывающего лишая среди лиц в возрасте 50 лет и старше [2]. А затем, в конце июля 2021-го, — для такой же задачи среди лиц в возрасте 18 лет и старше, находящихся в группе риска развития опоясывающего лишая ввиду иммунодефицита или иммуносупрессии по причине заболевания или терапии [3].
В Европе «Шингрикс» заручилась аналогичными маркетинговыми разрешениями Европейского агентства по лекарственным средствам (EMA) в конце марта 2018 года и конце марта 2020-го [4].
В России «Шингрикс» прошла регистрацию Минздрава по указанным двум показаниям в конце августа 2023 года [5].
БОЛЬШИЕ ДЕНЬГИ
Если в 2022 году «ГлаксоСмитКляйн» (GlaxoSmithKline) наторговала вакциной «Шингрикс» (Shingrix, RZV) на 2,96 млрд фунтов (3,67 млрд долларов), то в 2023-м ее реализация составила 3,45 млрд фунтов (4,27 млрд долларов).
[membership level=»2″ show_noaccess=»true»]
Перешагнуть планку в 1 млрд долларов продаж «Шингрикс» сумела мгновенно: через год после своего запуска.
Впечатляющий спрос на «Шингрикс» не идет ни в какое сравнение со скромными успехами вакцины «Зоставакс» (Zostavax, ZVL), которая, дебютировав в мае 2006 года, так и не смогла стать бестселлером: пик интереса пришелся на 2014 год, когда она принесла «Мерк и Ко» (Merck & Co.) 765 млн долларов.
Ввиду категорически превосходящей эффективности «Шингрикс» закат «Зоставакс» был неминуем. В середине ноября 2020 года «Мерк и Ко» прекратила ее продвижение в США, на главном рынке сбыта любой фармацевтической продукции [1].
История имеет и обратную сторону. В свое время «Мерк и Ко» и ее «Гардасил» / «Гардасил 9» (Gardasil / Gardasil 9) вытеснили «ГлаксоСмитКляйн» и ее «Церварикс» (Cervarix) из американского бизнеса прививок против вируса папилломы человека (ВПЧ). Так, если в 2023 году мировые продажи первых вышли к внушительным 3,94 млрд долларов, то второй — к скромным 117 млн фунтов (145 млн долларов).
[/membership]
ЭФФЕКТИВНОСТЬ
Клинические исследования ZOE-50 (ZOSTER-006, NCT01165177) и ZOE-70 (ZOSTER-022, NCT01165229) фазы III (рандомизированные, двойные слепые, плацебо-контролируемые, многоцентровые, международные), привлекшие свыше 31 тыс. человек соответственно в возрасте ≥ 50 и ≥ 70 лет, установили, после наблюдений в течение медианных 3,1 и 4,0 лет, следующую высокую профилактическую эффективность «Шингрикс» (Shingrix, RZV) против опоясывающего лишая [1] [2] [3] [4]:
- ≥ 50 лет: 97% (95% ДИ [здесь и далее]: 94–99);
- ≥ 60 лет: 98% (93–100);
- ≥ 70 лет: 91% (87–95);
- ≥ 80 лет: 91% (80–97).
«Шингрикс» аналогично успешно предотвратила развитие постгерпетической невралгии:
- ≥ 50 лет: 100% (77–100);
- ≥ 60 лет: 100% (41–100);
- ≥ 70 лет: 89% (69–97);
- ≥ 80 лет: 71% (−52–97).
«Шингрикс» защитила от развития осложнений опоясывающего лишая, отличных от постгерпетической невралгии, таких как васкулит, инсульт, а также диссеминированное, офтальмологическое, неврологическое или висцеральное заболевание. Профилактическая эффективность составила 94% (60–100) и 92% (43–100) среди лиц в возрасте ≥ 50 и ≥ 70 лет соответственно [5].
[membership level=»3″ show_noaccess=»true»]
«Шингрикс» надежно предотвращала развитие опоясывающего лишая на протяжении длительного времени после иммунизации, успешно преодолевая проблему старения иммунной системы и обеспечивая стойкую защиту вне зависимости от возраста. Так, после иммунизации «Шингрикс» превентивный щит поддерживался как минимум в течение 3 лет после прививки лиц в возрасте ≥ 50 лет [6] [7]. Эффективность вакцины на протяжении медианных 7 лет составила 91% (88–93), притом что уровень защиты изменялся следующим образом: 98% → 93% → 92% → 90% → 85% → 85% → 84% [8].
- Для сравнения: эффективность вакцины «Зоставакс» (Zostavax, ZVL) для защиты от опоясывающего герпеса лиц в возрасте ≥ 50 лет ослабевала быстро, составляя 68% (65–70) в первый год после прививки, 47% (44–50) в течение второго года и 32% (15–45) по прошествии восьми лет [9].
Долгосрочные наблюдения за участниками (n>7000) клинического испытания ZOSTER-049 (NCT05371080) фазы III (рандомизированного, открытого, многоцентрового, международного) выяснили, что после вакцинации «Шингрикс» иммунитет против опоясывающего герпеса сохранялся очень долгое время на прилично высоком уровне [10]. Показатели устойчивой защитной эффективности таковы:
- ≥ 50 лет: 80% (74–85) — в период от 6 до 11 лет после прививки;
- ≥ 50 лет: 82% (63–92) — на 11-м году после вакцинации;
- ≥ 70 лет: 73% (63–81) — в период от 6 до 11 лет после прививки.
Клинические исследования ZOSTER-002 (NCT01610414; #1) и ZOSTER-039 (NCT01767467; #2) фазы III (рандомизированные, двойные слепые, плацебо-контролируемые, многоцентровые, международные) пригласили взрослых (18 лет и старше) пациентов (n=1721 и n=515) с ослабленной иммунной системой (иммунокомпрометированным статусом), — соответственно прошедших аутологичную трансплантацию гемопоэтических клеток или с гематологическими злокачественными новообразованиями (множественная миелома, неходжкинская B-клеточная лимфома, хронический лимфоцитарный лейкоз и др.) [11] [12].
Защитная эффективность «Шингрикс» составила:
- ≥ 18 лет (#1): 68% (55–78);
- ≥ 50 лет (#1): 67% (53–78);
- ≥ 18 лет (#2): 87% (44–99).
Эффективность «Шингрикс» в задаче предупреждения постгерпетической невралгии следующая:
- ≥ 18 лет (#1): 89% (23–100);
- ≥ 50 лет (#1): 88% (10–100).


Клиническое исследование NCT02581410 фазы III (нерандомизированное, открытое, многоцентровое) подтвердило, что «Шингрикс» успешно справляется с профилактикой опоясывающего лишая среди лиц, ранее привитых альтернативной вакциной «Зоставакс» [13] [14].
Клинические исследования NCT01954251, NCT02045836, NCT03439657 и NCT02052596 фазы III (рандомизированные, открытые, многоцентровые, многоцентровые, международные) продемонстрировали, что «Шингрикс» можно без каких-либо проблем назначать совместно с другими вакцинами, такими как противогриппозная «Флуарикс» (Fluarix), пневмококковые «Пневмовакс 23» (Pneumovax 23) и «Превенар 13» (Prevnar 13), а также «Бустрикс» (Boostrix) против столбняка, дифтерии и коклюша — не обнаружено признаков взаимного вмешательства в иммунный ответ со стороны антигенов, содержащихся в этих вакцинах [15] [16] [17] [18]. Параллельную вакцинацию уместно осуществлять инъекциями в разные анатомические места [19] [20].
Клиническое исследование ZOSTER-026 (NCT01751165) фазы III (рандомизированное, открытое, многоцентровое, международное) показало, что вторую (бустерную) дозу «Шингрикс» можно вводить равно как через 2 месяца после первой, так и по прошествии 6 месяцев [21].
[/membership]
АЛЬТЕРНАТИВЫ
С октября 2017 года в Южной Корее доступна вакцина «Скайзостер» (SKYZoster), разработанная местной «Эс-кей байосайенсиз» (SK Bioscience) для профилактики опоясывающего лишая у лиц в возрасте 50 лет и старше [1].
[membership level=»2,3″ show_noaccess=»true»]
Защитная эффективность живой аттенуированной вакцины «Скайзостер» (NBP608) находится на приблизительно таком же уровне, как у «Зоставакс» (Zostavax, ZVL) авторства «Мерк и Ко» (Merck & Co.) [2].
В 2022 году «Скайзостер» занимала 54% корейского рынка, оставшиеся 46% принадлежали «Зоставакс» [3].
Однако в 2023-м, в первый год доступности «Шингрикс» (Shingrix, RZV), всё резко поменялось: вакцина «ГлаксоСмитКляйн» (GlaxoSmithKline) мгновенно захватила 44% продаж, оставив «Скайзостер» и «Зоставакс» по 30% и 26%. Что примечательно, клиническому внедрению «Шингрикс» не помешала ее относительно высокая цена в 500 тыс. вон (365 долларов) — против стоимости «Скайзостер» в 150 тыс. вон (110 долларов) и «Зоставакс» в 170 тыс. вон (125 долларов) [4].
В январе 2023 года Китай одобрил первую вакцину для профилактики опоясывающего лишая собственной разработки. Живая аттенуированная вакцина создана «Чанчунь Би-си-эйч-ти байотекнолоджи» (Changchun BCHT Biotechnology) [5].
[/membership]
ТЕМ ВРЕМЕНЕМ
В начале 2024 года «Курево ваксин» (Curevo Vaccine), за которой стоит корейская «Си-джи байофарма» (CG Biopharma), уведомила об успешной клинической проверке амезосватеина (amezosvatein, CRV-101), экспериментальной адъювантной субъединичной вакцины на основе гликопротеина E (gE) вируса ветряной оспы, которая прошла прямое сравнение с «Шингрикс» (Shingrix, RZV) [1].
[membership level=»2,3″ show_noaccess=»true»]
В клиническом исследовании NCT05304351 фазы II вакцинный кандидат амезосватеин продемонстрировал равно как нехудшую профилактическую эффективность против опоясывающего лишая, так и улучшенный профиль безопасности.
Как утверждает «Курево», несмотря на высокий уровень защиты от опоясывающего герпеса, которым наделяет «Шингрикс», степень ее внедрения среди нуждающихся в этой прививке лиц остается низкой. Так, заявлено, что две трети американцев не иммунизированы против опоясывающего герпеса, тогда как в Европе, Китае и в целом в мире «Шингрикс» получили менее 5% представителей группы риска развития опоясывающего лишая. Проблема связана в том числе с переносимостью вакцины авторства «ГлаксоСмитКляйн» (GlaxoSmithKline): якобы после первой дозы «Шингрикс» приблизительно 20–30% человек не обращаются за второй ее дозой по причине нежелательных явлений (НЯ).
Согласно исследованиям самой «ГлаксоСмитКляйн», действительно после «Шингрикс» широко распространены такие НЯ, как боль (в среднем в 80% случаев), покраснение (38%) или отечность (27%) в месте инъекции, боль в мышцах (47%), усталость (47%), головная боль (40%), озноб (29%), повышение температуры тела (22%), тошнота, рвота, диарея или боль в области живота (18%) [2]. Однако нежелательные явления не приводили к столь внушительному, как уверяет «Курево», проценту отказа от второй дозы «Шингрикс» [3] [4].
[/membership]
КОНВЕЙЕР
Фармпроизводители не оставляют надежду предложить новые профилактические вакцины, которые бы эффективнее, чем «Шингрикс» (Shingrix, RZV), справлялись с предупреждением опоясывающего лишая и его осложнений.
[membership level=»2,3″ show_noaccess=»true»]
В четвертом квартале 2024 года китайская «Бэйцзин Лучжу байотекнолоджи» (Beijing Luzhu Biotechnology) намеревается отправить на регистрацию LZ901 — рекомбинантную вакцину, сделанную на основе гликопротеина E (gE) вируса ветряной оспы в тетрамерном исполнении и подкрепленную классическим адъювантным гидроксидом алюминия [1] [25].
Китайская «Цзянсу Рекбайо текнолоджи» (Jiangsu Recbio Technology) работает над REC610 — рекомбинантной gE-вакциной с фирменным адъювантом BFA01, который позиционируется усовершенствованной версией адъюванта AS01B в «Шингрикс» [2] [3] [4] [5].
Китайская «Кансино байолоджикс» (CanSino Biologics) совместно с британской «Баринтус байотерапьютикс» (Barinthus Biotherapeutics), ранее называвшей «Вакситек» (Vaccitech), трудится над VTP-400 (ChAdOx1-VZV, CS-2032, CSB-016) — вакциной на базе аденовирусного вектора шимпанзе (ChAdOx1), кодирующего антиген gE [6].
Китайская «Имморна байотекнолоджи» (Immorna Biotechnology) верит в успех JCXH-105 — вакцины на базе самореплицирующейся РНК (срРНК), кодирующей антиген gE [7] [8] [9].
Китайская «Максвакс байотекнолоджи» (Maxvax Biotechnology) исследует рекомбинантную gE-вакцину с фирменным адъювантом MA105, составленным из полиинозиновой:полицитидиловой кислоты (Poly I:C), функционирующей как агонист толл-подобного рецептора 3 (TLR3), молекулы сапонина (QS-21) из растительного экстракта квиллайи мыльной (Quillaja saponaria), липидных молекул [10] [11]
Корейская «Си-эйч-эй ваксин рисёрч инститьют» (CHA Vaccine Research Institute) изучает CVI-VZV-001 — рекомбинантную gE-вакцину с фирменным адъювантом L-pampo, который представляет собой агонист толл-подобных рецепторов 1/2 и 3 (TLR1/2 и TLR3) [12] [13] [14].
Корейская «Эубайолоджикс» (EuBiologics) сделала ставку на рекомбинантную вакцину, нанолипосомальные частицы которой демонстрируют иммунной системе антиген gE наряду с адъювантами EcML и CoPoP, представляющими собой соответственно монофосфорил-липид A (MPL) кишечной палочки (Escherichia coli) и кобальт-порфирин-фосфолипид (CoPoP) [15].
Корейская «ДженУан лайф сайенс» (GeneOne Life Science) прорабатывает ДНК-вакцину GLS-5100 (VGX-5100). Лежащая в ее основе ДНК-плазмида кодирует гликопротеин E (gE) и предранние белки IE63 и IE62 вируса ветряной оспы, а также адъюванты интерлейкин 7 (IL-7) и интерлейкин 33 (IL-33) [16] [17].
Корейская «Айджин» (EyeGene) исследует EZ-HZ — рекомбинантную gE-вакцину с адъювантом CIA05, агонистом толл-подобного рецептора 4 (TLR4) [18].
«Дайнавакс текнолоджис» (Dynavax Technologies) занимается клинической проверкой Z-1018 — рекомбинантной gE-вакцины, подкрепленной адъювантом CpG 1018, который является агонистом толл-подобного рецептора 9 (TLR9), и опциональным гидроксидом алюминия [19].
Не забыта, разумеется, мРНК-технология: «Байонтек» (BioNTech) и «Пфайзер» (Pfizer) тестируют BNT167, «Модерна» (Moderna) пробует силы с mRNA-1468, академические учреждения Китая разработали «Зосал» (Zosal) и mgE@Syn-LNP. Все эти вакцины против опоясывающего лишая кодируют антиген gE [20] [21] [22] [23] [24].
[/membership]