ЧТО ПРОИЗОШЛО
«Сайтокинетикс» (Cytokinetics) разработала афикамтен (aficamten) — новый лекарственный препарат, предназначенный для лечения симптоматической обструктивной гипертрофической кардиомиопатии.
ОСНОВНЫЕ ФАКТЫ
Гипертрофическая кардиомиопатия (ГКМП) — одно из самых распространенных наследственных заболеваний сердца, вызывающее одышку при нагрузке и снижение физической работоспособности, что ухудшает качество жизни.
Афикамтен, пероральный ингибитор сердечного миозина, успешно прошел итоговую клиническую проверку: он улучшил способность к повседневной физической деятельности и облегчил симптомы обструктивной ГКМП.
В текущем 2024 году «Сайтокинетикс» отправит регистрационное досье афикамтена в адрес регуляторов. На рынок препарат поступит в 2025 году.
Прямым конкурентом афикамтена является «Камзайос» (Camzyos, мавакамтен), в конце апреля 2022 года предложенный «Бристол-Майерс Сквибб» (Bristol-Myers Squibb) и ставший первым в мире препаратом для специфического лечения симптоматической обструктивной ГКМП.
Несмотря на то что афикамтен характеризуется рядом преимуществ перед мавакамтеном (mavacamten), будущий спрос на него упирается в ценовую политику, которую изберет «Сайтокинетикс».

«Камзайос»: первое специфическое лекарство против гипертрофической кардиомиопатии
Мавакамтен, разработанный Bristol-Myers Squibb, — новый вздох для всех больных обструктивной гипертрофической кардиомиопатией.
ПРЯМАЯ РЕЧЬ
«Ингибирование сердечного миозина — отличный вариант терапии для пациентов с симптоматической обструктивной гипертрофической кардиомиопатией. Афикамтен, клинически значимым образом улучшающий способность к физической нагрузке, причем без событий с низкой фракцией выброса левого желудочка, когда приходится прерывать лечение, станет желанным дополнением для пациентов и врачей».
Мартин Мэрон (Martin Maron), ведущий автор исследования, директор Центра гипертрофической кардиомиопатии при Больнице и медицинском центре Лэхи (LHMC, Берлингтон, шт. Массачусетс, США).
«Мы убедились, что терапевтический эффект афикамтена охватывает диапазон, связанный с полноценной повседневной активностью. Афикамтен, положительно влияя на сердечную функцию, повышает общую работоспособность при физических нагрузках, что тесно коррелирует с улучшением нескольких важных показателей заболевания, включая структуру и функцию сердца, симптоматику».
Грегори Льюис (Gregory Lewis), соавтор исследования, заведующий кафедрой и секцией сердечной недостаточности, директор лаборатории тестирования сердечно-легочной деятельности, медицинский директор программы трансплантации сердца Центральной больницы Массачусетса (Бостон, шт. Массачусетс, США).
«Фармакологические свойства, заложенные в афикамтене, похоже, воплотились в клинические преимущества, которые до завершения исследования можно было только прогнозировать. Применение афикамтена характеризовалось быстрым выходом к максимальному терапевтическому эффекту у большинства пациентов, а частота нежелательных явлений была схожа с плацебо. Афикамтен — многообещающее лекарственное средство, востребованное при обструктивной гипертрофической кардиомиопатии».
Кэролайн Коутс (Caroline Coats), соавтор исследования, ведущий клиницист Службы наследственных заболеваний сердца Западной Шотландии (Глазго, Великобритания).
«Результаты исследования полностью оправдали высокие ожидания в отношении эффективности и безопасности, продемонстрировав, что афикамтен, добавленный к стандартной терапии, оказал положительное влияние на физическую работоспособность, а также быстрое и устойчивое воздействие на симптомы и функциональный класс у пациентов с обструктивной гипертрофической кардиомиопатии при сохранении безопасности и переносимости».
Фади Малик (Fady Malik), исполнительный вице-президент разработок и исследований «Сайтокинетикс» (Cytokinetics).
«Афикамтен всецело подтвердил свою способность благотворного влияния на качество жизни при обструктивной гипертрофической кардиомиопатии. В случае низкой левожелудочковой фракции нет никаких проблем с тем, чтобы снизить дозу препарата, тем самым индивидуализировав его назначение в целях нивелирования рисков».
Стюарт Купфер (Stuart Kupfer), старший вице-президент, медицинский директор «Сайтокинетикс» (Cytokinetics).
СУТЬ ВОПРОСА
Гипертрофическая кардиомиопатия (ГКМП) — одно из самых распространенных наследственных сердечно-сосудистых заболеваний во всём мире, которым страдает приблизительно каждый второй на тысячу человек [1]. ГКМП, в равной степени поражающая мужчин и женщин, — гетерогенное заболевание с выраженным разнообразием клинических проявлений, естественного течения болезни и ее прогноза [2] [3] [4] [5] [6] [7] [8].
Гипертрофическая кардиомиопатия характеризуется утолщенным, недилатированным (нерасширенным) левым желудочком [2] [4] [7], часто вызывает одышку при нагрузке и снижение физической работоспособности, тем самым ухудшая качество жизни [4] [7] [9] [10].
У приблизительно 70% пациентов с фенотипической ГКМП выявляется обструкция выходного тракта левого желудочка (ВТЛЖ) [15]. Механизмы развития обструкции определены и включают в себя сложное взаимодействие во время систолы между изменениями желудочкового кровотока и асимметричной гипертрофией межжелудочковой перегородки и створками митрального клапана. Именно обструкция является одним из основных факторов, определяющих связанные с ГКМП осложнения, и поэтому выступает важной целью лечения [11] [12] [13].
Гиперконтрактильность (усиленная сократимость) сердца, которая возникает из-за чрезмерного количества актин-миозиновых поперечных мостиков в саркомерах миокарда, является ключевым механизмом, способствующим обструкции оттока. Другие факторы включают удлинение створок митрального клапана, апикальное смещение папиллярных мышц и выпячивание гипертрофированной межжелудочковой перегородки в ВТЛЖ [12] [14]. Препятствие кровотоку создает градиент давления в ВТЛЖ, который может быть надежно определен с помощью эхокардиографии [2] [4] [7] [11] [13].
ПОЧЕМУ ЭТО ВАЖНО
Лечение обструктивной гипертрофической кардиомиопатии (ГКМП) осуществляется как медикаментозно, так и инвазивно.
Фармакологические препараты для лечения обструктивной ГКМП представлены бета-блокаторами, блокаторами кальциевых каналов, дизопирамидом (disopyramide). Однако их эффективность ограничена, включая субоптимальное снижение градиента давления в выходном тракте левого желудочка (ВТЛЖ) и недостаточное облегчение симптомов. Не доказано, что данные лекарственные средства улучшают объективные показатели физической нагрузки. Их назначение приводит к определенным побочным эффектам, недопустимым у некоторых пациентов [1] [2] [3] [4] [5] [6].
Инвазивные методы лечения обструктивной ГКМП, показанные лицам с выраженными симптомами или функциональным дефицитом, такие как хирургическая миоэктомия и чрескожная спиртовая абляция межжелудочковой перегородки, эффективны для уменьшения градиента давления в ВТЛЖ и благоприятно влияют на клиническое течение, включая длительное облегчение ограничивающих физическую активность симптомов [7]. Однако эти вмешательства связаны с риском и обычно проводятся только в отдельных крупных хирургических центрах [8].
Современные стратегии лечения обструктивной ГКМП привели к тому, что большинство пациентов достигают продолжительности жизни, нормальной или близкой к ней, а показатели заболеваемости улучшились. Тем не менее по-прежнему высока потребность в новых лекарственных препаратах, которые были бы безопасными и надежно улучшали самочувствие и функционирование пациентов с минимальными побочными эффектами [2] [5].
КАК ЭТО РАБОТАЕТ
Гипертрофическая кардиомиопатия (ГКМП) возникает в результате патогенных генетических мутаций, затрагивающих гены, кодирующие белки сердечного саркомера, такие как миозин [1] [2]. Такие мутации, есть мнение, механистически увеличивают чистое производство энергии в саркомере in vitro [3] [4] [5] [6]. Результаты исследований согласуются с основной патофизиологией миокарда левого желудочка у пациентов с ГКМП: он характеризуется гиперконтрактильностью (сильным сокращением) со сниженным комплаенсом (растяжимостью) [7]. Всё это расширило понимание молекулярного патогенеза ГКМП и стимулировало усилия по поиску модуляторов сердечного миозина (моторного белка, управляющего сокращениями сердечной мышцы), способных воздействовать на основной механизм гиперконтрактильности при обструктивной гипертрофической кардиомиопатии [8].
Афикамтен (aficamten, CK-3773274, CK-274), разработанный «Сайтокинетикс» (Cytokinetics), представляет собой низкомолекулярный селективный обратимый аллостерический ингибитор сердечного миозина нового поколения. Афикамтен, посредством ингибирования активированной актином АТФазы в миозине с одной головкой и миозине с двумя головками, стабилизирует миозин в освобожденном состоянии после эффективного удара, делая невозможной гидролизацию АТФ [9] [10].

Афикамтен уменьшает количество активных актин-миозиновых поперечных мостиков во время каждого сердечного цикла, тем самым подавляя гиперконтрактильность миокарда, лежащей в основе патофизиологии гипертрофической кардиомиопатии. Предполагаемый фармакологический эффект афикамтена заключен в снижении силы, создаваемой саркомерами, что отражается уменьшением обструкции выходного тракта левого желудочка (ВТЛЖ) и улучшением диастолической функции. Снижение сократительной силы сердца должно облегчить обструкцию ВТЛЖ за счет противодействия чрезмерному утолщению левожелудочковой стенки в этом тракте [11] [12] [13].
Прямое ингибирование сердечного миозина для снятия гиперконтрактильности и обструкции ВТЛЖ при обструктивной ГКМП — это таргетный фармакологический подход, направленный на улучшение функциональных возможностей пациентов и симптомов их заболевания.
Весной 2022 года «Бристол-Майерс Сквибб» (Bristol-Myers Squibb) предложила первое такое лекарство в лице препарата «Камзайос» (Camzyos, мавакамтен), который улучшает способность к физической нагрузке и облегчает симптомы у пациентов с обструктивной ГКМП. Мавакамтен (mavacamten) является низкомолекулярным селективным аллостерическим ингибитором сердечной миозиновой АТФазы [14] [15] [16] [17].
Несмотря на схожий механизм действия, фармакокинетический профиль афикамтена несколько отличается от такового у мавакамтена. Так, период полувыведения мавакамтена составляет 7–9 дней, а равновесное состояние в плазме достигается приблизительно через 6 недель [14] [18]. У афикамтена существенно более короткий период полувыведения (где-то 3,4 дня), а на выход к равновесному состоянию уходит 2 недели, что позволяет быстрее подбирать его терапевтическую дозу и, в случае необходимости, быстрее отменять его действие путем снижения дозы [11].
КЛИНИЧЕСКИЕ ПОДРОБНОСТИ
Опорное клиническое исследование SEQUOIA-HCM (NCT05186818) фазы III (рандомизированное, двойное слепое, плацебо-контролируемое, многоцентровое, международное) пригласило взрослых пациентов (n=282) с симптоматической гипертрофической кардиомиопатией и обструкцией выходного тракта левого желудочка.
Среди основных требований к участникам:
- диагноз гипертрофической кардиомиопатии, подтвержденный левожелудочковой гипертрофией без расширения (дилатации) камеры левого желудочка при отсутствии другого кардиологического заболевания, с конечно-диастолической толщиной стенки левого желудочка либо ≥ 15 мм в как минимум одном миокардиальном сегменте, либо ≥ 13 мм в как минимум одном сегменте стенки и с известной генетической мутацией, вызывающей гипертрофическую кардиомиопатию, или семейным анамнезом последней;
- пиковый градиент давления выходного тракта левого желудочка (LVOT-G) в покое ≥ 30 мм рт. ст. и LVOT-G после пробы Вальсальвы ≥ 50 мм рт. ст.;
- фракция выброса левого желудочка (LVEF) ≥ 60%;
- хроническая сердечная недостаточность II или III функционального класса по NYHA;
- дыхательный коэффициент (RER) ≥ 1,05 и пиковое потребление кислорода (pVO2) ≤ 90%.
На протяжении 24 недель испытуемые ежедневно перорально получали плацебо или афикамтен (в дозе 5, 10, 15 или 20 мг). Разрешался фоновый прием уже назначенных кардиологических препаратов, таких как бета-блокаторы, верапамил, дилтиазем, дизопирамид.
Первичная конечная точка эффективности лечения обструктивной гипертрофической кардиомиопатии была установлена изменением пикового потребления кислорода (pVO2), согласно кардиопульмональному нагрузочному тесту (CPET).
Применение афикамтена привело к росту этого показателя на усредненных 1,8 (95% ДИ [здесь и далее]: 1,2–2,3) мл/кг/мин — против изменения на 0,0 (−0,5, 0,5) мл/кг/мин в группе плацебо. Расхождение получилось статистически значимым: среднеквадратичная средняя разница составила 1,7 (1,0–2,4) мл/кг/мин (p<0,001) [1].
Афикамтен обеспечил статистически значимые (p<0,0001) и клинически значимые улучшения всех десяти вторичных конечных точек, включая:
- суммарный балл клинических симптомов по Канзасскому опроснику для больных кардиомиопатией (KCCQ-CSS) после 12 и 24 недель терапии (W12 и W24): +11 в группе афикамтена — против +5 в группе плацебо;
- снижение функционального NYHA-класса хотя бы на один (W12 и W24): у 49% и 59% пациентов — против 18% и 24%;
- LVOT-G после выполнения физических упражнений (W12 и W24): −44,8 и −47,6 мм рт. ст. — против +2,8 и +1,8 мм рт. ст.;
- пропорцию пациентов с LVOT-G < 30 мм рт. ст. (W12 и W24): 52% и 49% — 6% и 4%;
- продолжительность статуса пригодности к септальной редукционной терапии на протяжении 24-недельного периода лечения: +37 дней — против +114 дней;
- изменение общей рабочей нагрузки, согласно CPET на 24-й неделе терапии: + 14 Вт — против +1 Вт.
Уровень N-концевого фрагмента мозгового натрийуретического пептида (NT-proBNP) после 24 недель лечения изменился на среднегеометрических пропорциональных +0,20 (0,17–0,22) пг/мл — против +1,00 (0,91–1,07) пг/мл.
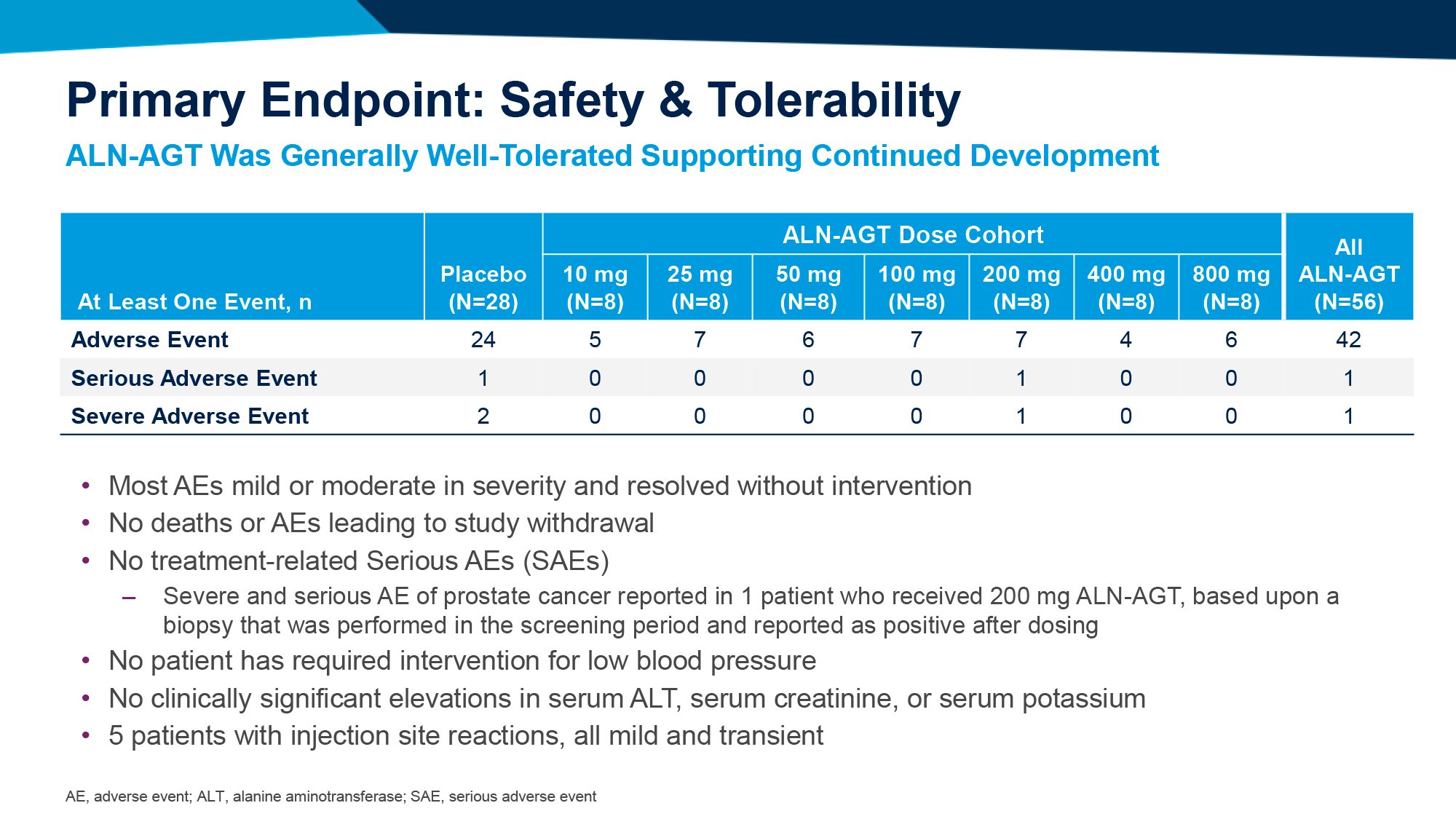
Афикамтен характеризовался приемлемой переносимостью: профиль нежелательных явлений (НЯ) был сравним с таковым у плацебо. Серьезные НЯ, вызванные лечением, были зарегистрированы у 5,6% пациентов в группе афикамтена — против 9,3% в группе плацебо.






ЧТО ЕЩЕ
Согласно промежуточным 48-недельным результатам непрерывного назначения афикамтена в FOREST-HCM (NCT04848506) обеспечило следующие терапевтически благотворные изменения в ходе лечения обструктивной гипертрофической кардиомиопатии (n=46) [1]:
- LVOT-G в покое −39,6 мм рт. ст.;
- LVOT-G по Вальсальве −53,2 мм рт. ст.;
- улучшение функционального NYHA-класса на ≥1 у 82%;
- снижение уровня NT-proBNP на 63%;
- улучшение показателей структуры и функции сердца, включая уменьшение максимальной толщины стенок, снижение индекса объема левого предсердия, уменьшение соотношения E/e′;
- снижение на 94% риска пригодности для септальной редукционной терапии.
Безопасность афикамтена не вызывала каких-либо претензий. Наблюдалось умеренной снижение левожелудочковой фракции выброса (−5,1 мг). Ввиду ее уменьшения ниже 50% трем пациентам потребовалась коррекция дозы афикамтена: у двух всё протекало бессимптомно, у третьего случился рецидив фибрилляции предсердий из-за алкоголя.
ЧТО ДАЛЬШЕ
В рамках клинической программы афикамтена «Сайтокинетикс» оценивает его потенциал как лекарственного средства, улучшающего способность к физической нагрузке и облегчающего симптомы у пациентов с гипертрофической кардиомиопатией, а также его долгосрочное влияние на структуру и функцию сердца. Для этого осуществляются следующие клинические испытания фазы II/III и III:
- MAPLE-HCM (NCT05767346): лечение афикамтеном симптоматической обструктивной гипертрофической кардиомиопатии у взрослых — сравнение с метопрололом;
- ACACIA-HCM (NCT06081894): лечение афикамтеном симптоматической необструктивной гипертрофической кардиомиопатии у взрослых;
- CEDAR-HCM (NCT06412666): лечение афикамтеном симптоматической обструктивной гипертрофической кардиомиопатии в педиатрической популяции (12–17 лет);
- FOREST-HCM (NCT04848506): продолжение назначения афикамтена сроком до 5 лет в целях выяснения его долгосрочных безопасности и эффективности среди участников других испытаний.
ЭКСПЕРТНЫЕ КОММЕНТАРИИ
Обеспеченное афикамтеном улучшение пикового потребления кислорода, согласно кардиопульмональному нагрузочному тесту, оказалось больше в сравнении с результатами применения препарата «Камзайос» (Camzyos, мавакамтен), продвигаемого «Бристол-Майерс Сквибб»: соответствующие разницы с плацебо составили 1,74 (1,04–2,44) мл/кг/мин и 1,4 (0,6–2,1) мл/кг/мин.
Благотворное действие афикамтена проявилось во всех предварительно заданных подгруппах пациентов, включая тех, кто следовал или нет фоновой терапии бета-блокаторами: соответствующие разницы с плацебо составили 1,6 (0,7–2,5) мл/кг/мин и 1,9 (0,8–3,1) мл/кг/мин — это открывает для афикамтена двери для монотерапевтического использования.
Поскольку период полувыведения у афикамтена укорочен по сравнению с таковым у мавакамтена, существуют весомые предпосылки, что подбор адекватной для пациента терапевтической дозы будет происходить быстрее.
Профиль безопасности афикамтена выглядит более приемлемым, нежели таковой у мавакамтена. Так, снижение фракции выброса левого желудочка до менее чем 50% было зарегистрировано у 3,5% пациентов, принимавших афикамтен (притом что никто не прекратил лечения по этой причине), — против 6% при назначении мавакамтена.
Более того, инструкция по медицинскому применению лекарственного препарата «Камзайос» снабжена «чернорамочным» предупреждением относительно рисков развития сердечной недостаточности или полной блокады функции желудочков, так как мавакамтен уменьшает систолическое сокращение.
БИЗНЕС
Когда в конце 2023 года «Сайтокинетикс» объявила об успешной итоговой клинической проверке афикамтена, на волне благостных известий ее биржевые котировки прибавили свыше 80%: рыночная капитализация фармацевтического предприятия превысила 8 млрд долларов [1].
Приблизительно тогда же витали слухи, будто игроки «Большой фармы» присматриваются к покупке «Сайтокинетикс»: приобретение якобы было интересно «АстраЗенека» (AstraZeneca), «Новартис» (Novartis) и «Джонсон энд Джонсон» (Johnson & Johnson). Затем, впрочем, это желание якобы пропало [2] [3] [4].
Согласно отраслевым прогнозам, афикамтен, если будет одобрен, способен выйти на уровень ежегодных продаж $3 млрд долларов к 2035 году.
«Камзайос» (Camzyos, мавакамтен), который «Бристол-Майерс Сквибб» получила благодаря поглощению «Майокардиа» (MyoKardia) в ноябре 2020 года за $13,1 млрд наличными [5], заработал в 2022 и 2023 гг. $24 млн и $231 млн. За первую половину 2024-го он принес $223 млн.
С учетом того, что для незастрахованных американских пациентов стоимость годового курса лечения обструктивной гипертрофической кардиомиопатии препаратом «Камзайос» составляет без малого $100 тыс. (без учета скидок и дисконтов) [6], «Сайтокинетикс», возможно, разумно сыграть на большей ценовой доступности афикамтена.
ПАЙПЛАЙН
ГЕННАЯ ТЕРАПИЯ
Гипертрофическая кардиомиопатия (ГКМП) вызывается мутациями в генах, кодирующих белки сократительного аппарата кардиомиоцитов, что приводит к дезорганизации, фиброзу и утолщению миоцитов, в результате чего возникает дисфункция желудочков. Выявлено свыше 450 мутаций в 20 белках, связанных с саркомерами и миофиламентами, с различной фенотипической выраженностью и тяжестью заболевания. Подавляющее большинство мутаций, определяющих развитие ГКМП, происходит либо в гене MYH7, который кодирует тяжелую цепь бета-миозина, либо в гене миозин-связывающего белка C (MYBPC3), что в совокупности составляет приблизительно две трети известных патологических мутаций [1] [2] [3].
На это и рассчитывает «Теная терапьютикс» (Tenaya Therapeutics), разрабатывающая генную терапию TN-201 для лечения взрослых и детей с ГКМП по причине мутаций в гене MYBPC3. Однократно внутривенно вводимый аденоассоциированный генно-терапевтический препарат доставляет в организм функциональную копию MYBPC3 в целях остановки прогрессирования заболевания и обращения вспять его последствий [4] [5] [6] [7] [8].
Запущено клиническое исследование MyPEAK-1 (NCT05836259) фазы Ib, изучающее TN-201 среди взрослых (n=15) с симптоматической необструктивной ГКМП. Первые результаты будут получены ближе к концу 2024 года.
«Ингибиторы сердечного миозина дали новую надежду пациентам, но они не воздействуют на основную причину гипертрофической кардиомиопатии. Однократная генная терапия это делает, причем она работает как при обструктивных, так и необструктивных формах заболевания; в случае последних пока не доказано, что ингибиторы сердечного миозина помогают».
Фараз Али (Faraz Ali), исполнительный директор « терапьютикс» (Tenaya Therapeutics).
МОДУЛЯЦИЯ САРКОМЕРА МИОКАРДА
«Эджвайз терапьютикс» (Edgewise Therapeutics) занимается EDG-7500 — первым в своем классе пероральным низкомолекулярным селективным модулятором саркомера миокарда, предназначенным для замедления скорости раннего сокращения, снижения диастолического напряжения, улучшения диастолического наполнения и устранения нарушений расслабления сердца, связанных с гипертрофической кардиомиопатией и другими заболеваниями диастолической дисфункции. Мишень EDG-7500, который не оказывает прямого ингибирующего воздействия на моторную головку сердечного миозина, пока остается загадкой [1].
Согласно доклиническим данных на животных моделях обструктивной и необструктивной ГКМП, экспериментальный EDG-7500 вызывает благоприятный ответ при низком риске снижения фракции выброса левого желудочка ниже нормы при всех испытанных дозах. EDG-7500 характеризуется самоограничивающимся эффектом на систолическое сокращение, что позволяет применять его в фиксированной дозе без интенсивного мониторинга безопасности пациентов [2] [3] [4] [5] [6] [7] [8].
Продолжается клиническое исследование CIRRUS-HCM (NCT06347159) фазы II среди взрослых (n=55) с обструктивной и необструктивной ГКМП [9].
«Мы предполагаем, что EDG-7500 продемонстрирует мощное снижение градиента давления выходного тракта левого желудочка при сохранении его нормальной сократимости, а также обеспечит улучшение наполнения желудочков и диастолическую функцию».
Марк Семигран (Marc Semigran), директор по развитию «Эджвайз терапьютикс» (Edgewise Therapeutics).
ВОССТАНОВЛЕНИЕ ЭНЕРГЕТИЧЕСКОГО ГОМЕОСТАЗА МИОКАРДА
«Имбрия терапьютикс» (Imbria Pharmaceuticals) успешно провела препарат-кандидат нинерафаксстат (ninerafaxstat, IMB-101, IMB-1018972) через клиническое исследование IMPROVE-HCM (NCT04826185) фазы II среди взрослых (n=67) с симптоматической необструктивной гипертрофической кардиомиопатией и готовится к запуску FORTITUDE-HCM фазы III [1] [2] [3].
Нинерафаксстат — пероральный низкомолекулярный сердечный митотроп, предназначенный для восстановления энергетического гомеостаза миокарда [4] [5] [6]. Основной механизм действия нинерафаксстата заключен в частичном ингибировании окисления жирных кислот путем прямого конкурентного ингибирования тиолазы I (3-кетоацил-коэнзим-А-тиолаза, 3-KAT) — последнего фермента в митохондриальном пути бета-окисления длинноцепочечных жирных кислот [4] [5] [7]. Это приводит к смещению энергетического метаболизма сердца с окисления свободных жирных кислот на окисление глюкозы, уменьшая количество кислорода, необходимого на моль вырабатываемого аденозинтрифосфата (АТФ), и тем самым повышая эффективность работы сердца, особенно при ограниченном поступлении кислорода. Способность нинерафаксстата оптимизировать энергетическую эффективность сердца может привести к улучшению функции при физической нагрузке за счет улучшения расслабления, наполнения и ударного объема миокарда во время нагрузки [6].
«Улучшения, наблюдаемые при использовании нинерафаксстата как в отношении симптомов, так и функциональных возможностей пациентов с необструктивной гипертрофической кардиомиопатией, для которых нет одобренных вариантов лечения, явно поддерживает дальнейшую клиническую оценку этого препарата».
Мартин Мэрон (Martin Maron), ведущий автор исследования, директор Центра гипертрофической кардиомиопатии при Больнице и медицинском центре Лэхи (LHMC, Берлингтон, шт. Массачусетс, США).
ВОССТАНОВЛЕНИЕ ГОМЕОСТАЗА МЕДИ
Академические учреждения, возглавляемые Манчестерским университетом (Великобритания), пробуют, на рельсах клинического испытания TEMPEST (NCT04706429) фазы II, перепрофилировать триентин (trientine), используемый в лечении болезни Вильсона хелатирующий медь II агент, направив его против гипертрофической кардиомиопатии у взрослых (n=154) [1] [2]
Идея состоит в том, что при ГКМП нарушен гомеостаз меди (повышен уровень меди и церулоплазмина в сыворотке крови), а несвязанные и слабосвязанные тканевые ионы меди II являются мощными катализаторами реактивных форм кислорода (ROS) и окислительного стресса, а также ингибиторами ферментативных антиоксидантов, таких как внеклеточная супероксиддисмутаза (SOD). Триентин, удаляющий ионы меди II из тканей, ассоциирован с восстановлением ультраструктуры митохондрий и нормализацией экспрессии и ферментативной активности белков, участвующих в энергетическом обмене, компонентов митохондриальной дыхательной цепи и ферментов, участвующих в окислении жирных кислот [3] [4] [5] [6] [7] [8] [9] [10] [11].
Это потенциально очень важно в случае ГКМП, поскольку широко распространена гипотеза, что энергетическое истощение является механизмом, посредством которого генные мутации приводят к фенотипу заболевания [12]. У носителей генов с мутациями саркомеров наблюдается значительно ухудшенная энергетика миокарда еще до развития левожелудочковой гипертрофии, то есть, как предполагается, дефицит энергии является первичным событием [13]. Нарушение энергетики миокарда при ГКМП связано с прогрессирующим фиброзом миокарда [14]. Более того, наследственные дефекты в производстве энергии митохондриями и окислении жирных кислот приводят к фенотипам, имитирующим ГКМП [15]. Триентин также нормализует внеклеточную SOD, что подавляет ROS-опосредованную активацию тканевого фактора роста бета (TGF-β) и обращает фиброз миокарда [4] [5] [6].
Ввиду определенных проблемы с безопасностью хелаторов тяжелых металлов, включая нейротоксичность и гепатотоксичность, некоторые склоняются к использованию природных хелатирующих агентов, например куркумина [16].