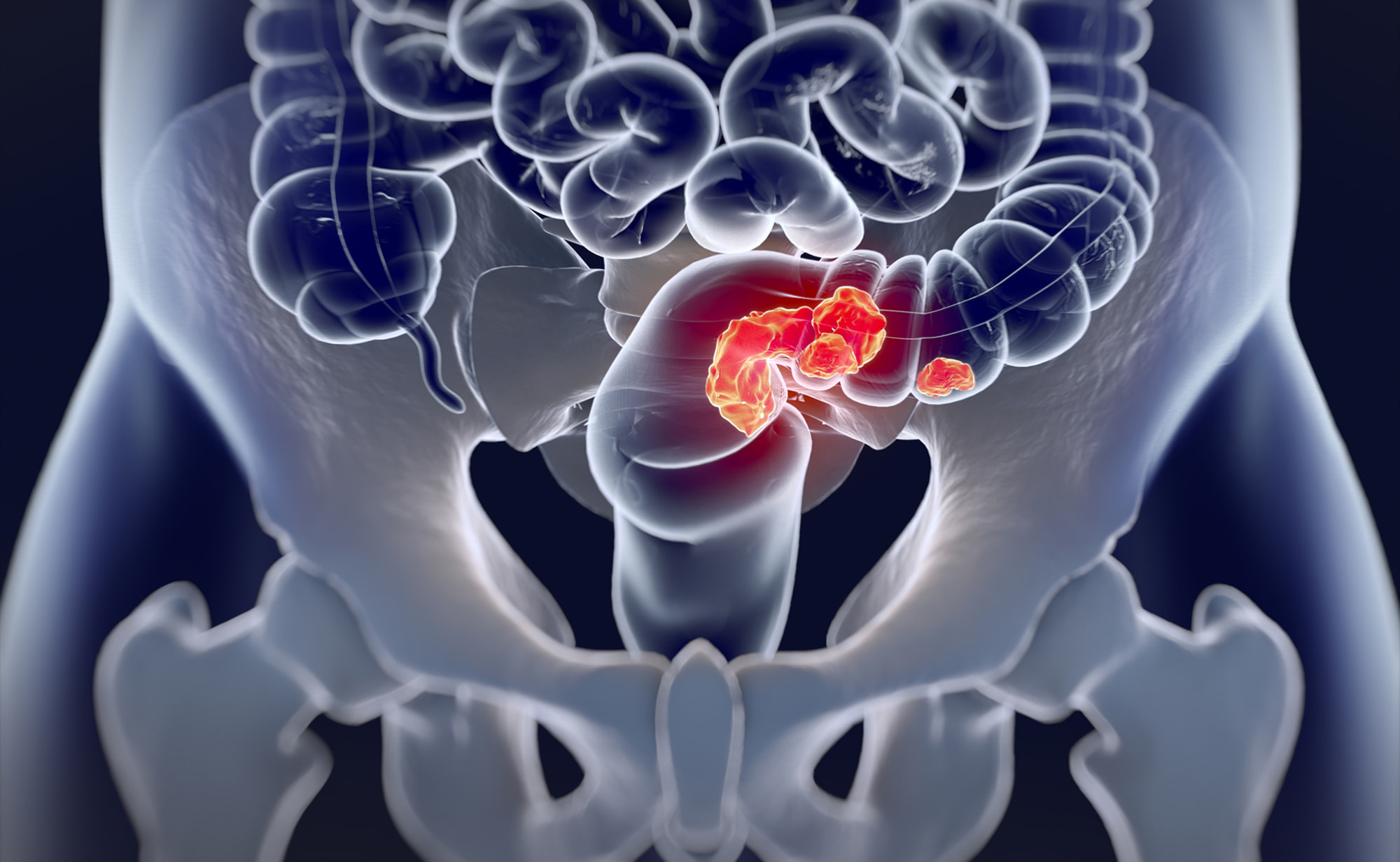
«Джемперли» (Jemperli, достарлимаб) обеспечил прежде невиданную ремиссию в ходе лечения рака прямой кишки.
ОСНОВНЫЕ ФАКТЫ
Шестимесячная монотерапия достарлимабом (dostarlimab) местнораспространенного рака прямой кишки с высокочастотной микросателлитной нестабильностью (MSI-H) или дефектами в системе репарации ошибочно спаренных оснований ДНК (dMMR) привела к тому, что после ее завершения больше половины пациентов, для которых были собраны клинические данные, вышли к стойкой ремиссии, которая продолжалась два года.
Никому из ответивших на экспериментальное лечение испытуемых не потребовалось последующее стандартное лечение.
«Джемперли» (Jemperli, достарлимаб), блокатор PD-1, предлагаемый «ГлаксоСмитКляйн» (GlaxoSmithKline), одобрен в лечении рецидивирующих или распространенных солидных опухолей с dMMR/MSI-H, включая рак эндометрия, идущий по отдельному терапевтическому показанию.
Иммунотерапевтический препарат достарлимаб продлит жизнь при распространенном или рецидивирующем раке эндометрия.
ПРЯМАЯ РЕЧЬ
«Достарлимаб, обеспечивший полную ремиссию на протяжении двух лет, должен стать совершенно новым подходом к лечению местнораспространенного рака прямой кишки с MSI-H/dMMR, не требующего стандартного вмешательства с его изнурительными для пациента последствиями».
Андреа Сёрсек (Andrea Cercek), заведующая отделением колоректального рака и содиректор Центра колоректального и желудочно-кишечного рака с ранним началом Мемориального онкологического центра им. Слоуна — Кеттеринга (MSK, Нью-Йорк, США).
«Собранные клинические данные приближают нас к пониманию потенциала достарлимаба в условиях местнораспространенного рака прямой кишки с MSI-H/dMMR. Беспрецедентная 100-процентная частота клинически полных ответов подтверждает грядущее изменение парадигмы лечения этой формы онкологии».
Хешам Абдулла (Hesham Abdullah), старший вице-президент и руководитель глобальных онкологических исследований и разработок «ГлаксоСмитКляйн» (GlaxoSmithKline).
СУТЬ ВОПРОСА
Рак прямой кишки — это форма рака, которая возникает в конечном отделе толстого кишечника и которая обычно относится к группе онкологических заболеваний, называемых колоректальным раком.
Колоректальный рак занимает третье место в мире по частоте диагностирования [1]. Приблизительно в 5–10% случаев рака прямой кишки отмечаются опухолевые аномалии dMMR/MSI-H, влияющие на корректность восстановления ДНК при ее копировании в клетке [2]. Такие аномалии являются биомаркером, который предсказывает успешный ответ на терапевтическую PD-(L)1-блокаду иммунных контрольных точек [3] [4]. Подобные опухоли чаще всего встречаются при раке эндометрия, колоректальном раке и других желудочно-кишечных онкологических заболеваниях, но могут обнаруживаться и при иных солидных опухолях [5] [6] [7] [8].
ПОЧЕМУ ЭТО ВАЖНО
Нынешний стандарт лечения местнораспространенного рака прямой кишки с MSI-H обращается к первоочередному назначению химиотерапии с облучением, затем проводится опухолевая резекция вместе с участками кишечника и/или окружающих тканей [1]. Это приводит к положительным результатам для большинства пациентов, однако почти одна треть больных в конечном итоге умирает ввиду отдаленного метастазирования [2]. Кроме того, оперативное вмешательство и химиорадиотерапия могут отражаться долгосрочными неблагоприятными последствиями, оказывающими серьезное негативное влияние на качество жизни по причине нарушения функции кишечника и мочеиспускания, половой дисфункции, вторичных раковых заболеваний, бесплодия [1].
КЛИНИЧЕСКИЕ ПОДРОБНОСТИ
Клиническое исследование NCT04165772 фазы II (нерандомизированное, открытое, многоцентровое) привлекло взрослых пациентов (n=16) с прежде нелеченной местнораспространенной ректальной аденокарциномой на стадии II/III с MSI-H/dMMR.
Протокол испытания предполагал, что участники получат неоадъювантный (до хирургического вмешательства) достарлимаб (каждые 3 недели на протяжении 6 месяцев), после чего, в случае остаточного заболевания, будет проведена стандартная лучевая терапия с одновременным назначением химиотерапевтического капецитабина, а затем, в случае сохранения заболевания, — тотальное мезоректальное иссечение.
По прошествии медианных 12 месяцев (6–25) наблюдений за пациентами (n=12), которые прошли лечение достарлимабом, результаты получились следующими [1]:
клинически полный ответ (cCR) составил 100% (95% ДИ [здесь и далее]: 74–100);
опухоль не обнаруживалась никаким из доступных способов, включая МРТ-изображения, ПЭТ-изображения, эндоскопию, пальцевое ректальное исследование, биопсию;
прогрессирования или рецидива заболевания не зафиксировано;
никому из пациентов не потребовались последующие химиорадиотерапия или хирургическое вмешательство.
[su_spoiler class=»my-custom-spoiler» title=»Эволюция эндоскопического и радиографического ответа у пациентов с раком прямой кишки, получавших лечение достарлимабом (dostarlimab). Предупреждение: медицинские изображения могут показаться неприемлемыми.»]
[/su_spoiler]
Столь впечатляющие результаты заставили «ГлаксоСмитКляйн» расширить набор пациентов, чтобы убедиться в высокой эффективности достарлимаба, и она не прогадала.
Так, по прошествии медианных 17,9 месяца (0,3–50,5) наблюдений за испытуемыми (n=42), которые полностью прошли 6-месячный курс лечения при помощи «Джемперли», cCR был зарегистрирован для всех 100% человек [2] [3].
Кроме того, по истечении медианных 26,3 месяца (12,4–50,5) наблюдений, все 24 пациента, для которых был доступен анализ данных, оставались в статусе устойчивого cCR, продолжавшегося как минимум 12 месяцев.
В целом медианное время до фиксирования клинически полного ответа составило 6,22 месяца (6,18–6,45).
ЧТО ДАЛЬШЕ
Продолжается клиническое испытание AZUR-1 (NCT05723562) фазы II, призванное подтвердить успехи достарлимаба в монотерапии прежде нелеченного местнораспространенного рака прямой кишки на стадии II/III (T3–T4, N0 или Tx, N+) с MSI-H/dMMR. Собираются данные, касающиеся пропорции пациентов, сохраняющих статус полной ремиссии по прошествии 12, 24 и 36 месяцев после лечения.
Осуществляется также клиническое исследование AZUR-2 (NCT05855200) фазы III, сравнивающее достарлимаб с химиотерапией в первоочередном лечении колоректального рака на стадии II (T4N0) или стадии III (операбельного) с MSI-H/dMMR. Накапливаются данные по частоте бессобытийной выживаемости (EFS) на протяжении 5 лет после терапии.
ЭКСПЕРТНЫЕ КОММЕНТАРИИ
Победа индукционного неоадъювантного назначения достарлимаба над раком прямой кишки обнадеживает. Облучение и хирургическое вмешательство имеют необратимые последствия для фертильности, сексуального здоровья, функции кишечника и мочевого пузыря [1] [2] [3] [4] [5]. Существенны также последствия для качества жизни, что особенно актуально для молодых людей детородного возраста, поскольку среди них растет заболеваемость раком прямой кишки [6].
Следует, впрочем, понимать, что аденокарцинома прямой кишки только в 5–10% случаев характеризуется наличием MSI-H/dMMR. Такие опухоли весьма плохо отвечают на стандартные схемы химиотерапии, включая неоадъювантную [7] [8] [9].
Важный вопрос заключен в том, почему локализованные опухоли прямой кишки с MSI-H/dMMR реагируют на имммуноонкологическое лечение гораздо сильнее, чем метастатические колоректальные опухоли с MSI-H/dMMR [10].
Одно из объяснений заключается в иммуномодулирующем влиянии микробиома кишечника: некоторые виды бактерий усиливают противоопухолевый иммунный ответ, потенцированный неоадъювантным назначением ингибитора иммунных контрольных точек (ИИКТ) [11] [12] [13]. В ряде клинических испытаний ИИКТ в лечении немелкоклеточного рака легкого (НМРЛ), меланомы и почечно-клеточной карциномы было подтверждено существенное благотворное влияние определенных бактерий кишечного микробиома на противоопухолевый ответ [14] [15] [16].
Кроме того, различия в ответах могут быть связаны с характеристиками опухолевых клеток помимо чрезвычайно высокого мутационного бремени (ввиду нарушенной системы репарации), такими как клональность, анеуплоидия и класс мутации [17] [18] [19].
Если разбивать медианное время до регистрации клинически полного ответа, согласно МРТ прямой кишки, эндоскопии, биопсии, уровню циркулирующей опухолевой ДНК (ctDNA) или ПЭТ-КТ, то оно получилось соответственно равным 6,15 месяца (6,09–6,25), 6,18 месяца (3,62–6,22), 1,41 месяца (1,38–2,73), 1,38 месяца (1,38–2,76) и 2,76 месяца (2,73–6,18).
Получается, что наиболее прогностически ранним оказался высокочувствительный анализ на ctDNA: изначально она обнаруживалась у 97% пациентов, а уже через 6 недель лечения перестала выявляться у более половины участников.
Продолжение наблюдений за пациентами из NCT04165772, чтобы установить окончательную продолжительность ремиссии, позволит выяснить, избавляет ли неоадъюватное иммуноонкологическое лечение от необходимости в хирургической резекции в долгосрочной перспективе.
В будущем, возможно, неоадъюватная блокада PD-1 окажется востребованной в лечении других опухолей с MSI-H/dMMR, таких как локализованный рак поджелудочной железы, желудка, предстательной железы. Следует подтвердить гипотезу востребованности высокоактивной противораковой терапии в неоадъювантном контексте до химиорадиотерапии и хирургического вмешательства, то есть до воздействия других агентов, которые могут нацелиться на клетки с резистентным фенотипом.
«Вертекс фармасьютикалс» (Vertex Pharmaceuticals) выпустила «Алифтрек» (Alyftrek) — новый препарат для лечения муковисцидоза, который эффективнее и удобнее в применении, чем «Трикафта» / «Кафтрио» (Trikafta / Kaftrio), сейчас позиционируемый наиболее совершенным противомуковисцидозным лекарственным средством.
ОСНОВНЫЕ ФАКТЫ
«Алифтрек» реализован следующей тройкой лекарственных средств, как то: ванзакафтор (vanzacaftor), тезакафтор (tezacaftor) и деутивакафтор (deutivacaftor) — они объединены в одной таблетке.
«Алифтрек» назначается один раз в день, тогда как «Трикафта» / «Кафтрио», представленный комбинацией из элексакафтора (elexacaftor), тезакафтора и ивакафтора (ivacaftor), — утром и вечером.
В конце декабря Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) одобрило «Алифтрек» в лечении муковисцидоза у пациентов в возрасте 6 лет и старше с хотя бы одной мутацией F508del или другой мутацией, чувствительной к назначению этого препарата [1].
Вердикт Европейского агентства по лекарственным средствам (EMA) аналогично не за горами.
Инструкция по медицинскому применению «Алифтрека», равно как «Трикафта» / «Кафтрио», снабжена «чернорамочным» предупреждением о рисках лекарственного поражения печени, потенциально опасного и жизнеугрожающего: в некоторых случаях это может привести к необходимости трансплантации печени или смертельному исходу. Перед началом лечения, а затем в его процессе следует отслеживать уровни печеночных ферментов.
Годовое лечение муковисцидоза при помощи «Алифтрека» обойдется американским пациентам в 370 тыс. долларов — стоимость «Трикафта» / «Кафтрио» чуть ниже: 346 тыс. долларов.
В 2023 году «Вертекс», пакет коммерциализированных препаратов которой был представлен только противомуковисцидозными лекарствами, заработала 9,9 млрд долларов, из которых 8,9 млрд долларов принес «Трикафта» / «Кафтрио».
Комбинированное лекарство поможет почти всем пациентам с муковисцидозом.
ПРЯМАЯ РЕЧЬ
«Дополнительное снижение уровня хлоридов в потовой жидкости, наблюдаемое при назначении новой тройной терапии муковисцидоза, определенно заслуживает внимания, поскольку выводит жизнь пациентов на качественно новый уровень».
Клэр Китинг (Claire Keating), содиректор программы по лечению муковисцидоза и легких у взрослых имени Гуннара Эсиасона (Gunnar Esiason Adult Cystic Fibrosis and Lung Program) в Медицинском центре имени Ирвинга при Колумбийском университете (Columbia University Irving Medical Center, CUIMC, Нью-Йорк, США).
«Я работаю детским пульмонологом уже более четырех десятилетий и на собственном опыте убедился, какое огромное влияние оказывают модуляторы CFTR на людей с муковисцидозом, превращая его из заболевания, укорачивающего жизнь, в нынешнее состояние, когда мы наблюдаем потенциал для остановки болезни еще до ее начала. Результаты новой тройной терапии муковисцидоза особенно поразительны в педиатрической популяции».
Бонни Рэмси (Bonnie Ramsey), старший консультант Сети разработки терапевтических препаратов (Therapeutics Development Network) Фонда муковисцидоза (Cystic Fibrosis Foundation, CFF, Бетесда, шт. Мэриленд, США) и сопредседатель руководящего комитета «Вертекс фармасьютикалс» (Vertex Pharmaceuticals) по CFTR-модуляторам.
«Ванзакафтор в составе новой тройной терапии муковисцидоза поднимает высокую планку, установленную препаратом „Трикафта“, предоставляя шанс большему количеству людей достичь уровня хлоридов в потовой жидкости не только ниже порога, необходимого для постановки диагноза муковисцидоза, но и даже ниже той концентрации, которая наблюдается у людей без муковисцидоза».
Ниа Тацис (Nia Tatsis), исполнительный вице-президент и главный специалист по регулированию и качеству «Вертекс фармасьютикалс» (Vertex Pharmaceuticals).
«За пять лет, прошедших с момента одобрения препарата „Трикафта“, лечение муковисцидоза претерпело значительные изменения, изменив перспективы пациентов. Наша новая тройная схема не уступает эффективности „Трикафта“ в улучшении функции легких и превосходит этот препарат в снижении концентрации хлоридов в потовой жидкости, тем самым устанавливая новый стандарт лечения. Мы продолжаем двигаться к достижению нашей основной цели — привести всех пациентов к состоянию нормального функционирования белка CFTR».
Кармен Бозич (Carmen Bozic), исполнительный вице-президент по глобальной разработке лекарственных средств и медицинским вопросам, медицинский директор «Вертекс фармасьютикалс» (Vertex Pharmaceuticals).
КАК ЭТО РАБОТАЕТ
Муковисцидоз — аутосомно-рецессивное мультисистемное заболевание, вызванное патогенными мутациями гена муковисцидозного трансмембранного регулятора проводимости (CFTR), который кодирует одноименный белковый канал-переносчик для хлоридов и бикарбонатов. Нарушение транспорта хлоридов приводит к образованию густых, вязких выделений в легких, поджелудочной железе, печени, кишечнике и репродуктивном тракте. Пациенты с муковисцидозом страдают от тяжелых респираторных заболеваний, проблем с желудочно-кишечным трактом, недостаточности поджелудочной железы, бесплодия [1] [2].
Мутации CFTR многочисленны (описано свыше 2,1 тыс. [3] [4]), и потому угодить с эффективным противомуковисцидозным лекарством каждому пациенту весьма затруднительно. Наиболее часто встречается (в приблизительно 90% случаев) мутация F508del, затрагивающая хотя бы один аллель CFTR [5] [6].
Мутация F508de относится к тому классу мутаций, которые приводят к неправильному сворачиванию CFTR-белка (мисфолдинг) с последующими его удержанием в эндоплазматическом ретикулуме и деградацией протеасомой. В результате неправильного внутриклеточного процессинга и нарушенной миграции на поверхности клетки оказывается недостаточное количество зрелых белков, обеспечивающих транспорт хлоридов.
Модуляторы CFTR — класс лекарственных препаратов, которые улучшают продуцирование, внутриклеточный процессинг и/или функции дефектного белка CFTR. Появление этих лекарств, которые корректируют, потенцируют, стабилизируют и/или амплифицируют CFTR, ознаменовало собой выдающийся прогресс в лечении муковисцидоза, поскольку они нацелены на непосредственно причину развития заболевания.
Самым продвинутым CFTR-модулятором является комбинированный препарат «Трикафта» / «Кафтрио» (Trikafta / Kaftrio, элексакафтор + тезакафтор + ивакафтор, ивакафтор), предложенный «Вертекс фармасьютикалс» (Vertex Pharmaceuticals) в конце октября 2019 года.
Ивакафтор (ivacaftor, VX-770), будучи CFTR-потенциатором (CFTR-стимулятором), увеличивает канальную активность расположенного на клеточной поверхности CFTR, что приводит к усилению транспорта хлоридов. Ивакафтор повышает вероятность открытия (гейтинга) ионного канала дефектного CFTR (путем продления времени удержания его в открытом состоянии), чтобы ионы хлора могли свободно пройти через него.
Тезакафтор (tezacaftor, VX-661), представляя собой CFTR-корректор, частично исправляет мисфолдинг CFTR (путем усиления его конформационной стабильности), тем самым обеспечивая рост количества CFTR на клеточной поверхности. Для успешной работы ивакафтора необходимо, чтобы CFTR располагался на клеточной поверхности, чему как раз и содействует тезакафтор.
Элексакафтор (elexacaftor, VX-445), который также является CFTR-корректором иначе модулирует CFTR, и потому его механизм действия вступает в синергизм с тезакафтором, обеспечивая дополнительное увеличение количества CFTR на клеточной поверхности.
Известно, что концентрация хлоридов в потовой жидкости является наиболее близким к точному показателем, отражающим транспортную функцию CFTR: снижение уровня хлоридов напрямую связано со снижением летальности и улучшением клинических исходов, таких как замедление скорости ухудшения легочной функции, уменьшение частоты трансплантации легких, улучшение физических параметров и роста [7] [8].
Хотя сочетание элексакафтора, тезакафтора и ивакафтора в лице комбинированного препарата «Трикафта» / «Кафтрио» улучшает функцию CFTR, что результирует обширным благотворным клиническим эффектом [9] [10] [11], лишь у небольшой части пациентов концентрация хлоридов в потовой жидкости достигает уровня у носителей муковисцидоза — людей с одной мутантной копией CFTR, у которых заболевание обычно протекает бессимптомно.
«Вертекс» вплотную взялась за решение этого вопроса, продолжив исследования в целях улучшения и модернизации лекарственных компонентов «Трикафта» / «Кафтрио» так, чтобы они обеспечивали более эффективный CFTR-опосредованный транспорт хлоридов. Итогом стало сочетание CFTR-потенциатора деутивакафтора (deutivacaftor, VX-561) и двух CFTR-корректоров — ванзакафтора (vanzacaftor, VX-121) и тезакафтора.
CFTR-потенциатор деутивакафтор — это ивакафтор, измененный химией дейтерия. Лекарственное соединение, приобретенное у «Кансет фармасьютикалс» (Concert Pharmaceuticals), более стабильно, и потому, в сравнении с оригинальным ивакафтором, характеризуется сниженной скоростью клиренса, повышенной экспозицией, более высокой концентрацией в плазме в течение 24 часов и более длительным периодом полувыведения [12] [13] [14].
CFTR-корректор ванзакафтор был выбран главным образом потому, что in vitro в сравнении с CFTR-корректором элексакафтором он приводил к увеличению транспорта хлоридов в несколько раз. Тройка из ванзакафтора, тезакафтора и деутивакафтора характеризовалась усиленными процессингом CFTR и CFTR-опосредованным транспортом хлоридов, если сравнивать с тройкой из элексакафтора, тезакафтора и ивакафтора [15] [16].
КЛИНИЧЕСКИЕ ПОДРОБНОСТИ
Опорная клиническая программа, которая изучила эффективность и безопасность противомуковисцидозной лекарственной комбинации «Алифтрек» (Alyftrek), составленной из ванзакафтора (vanzacaftor), тезакафтора (tezacaftor) и деутивакафтора (deutivacaftor), реализована тремя клиническими испытаниями фазы III:
SKYLINE 102 (NCT05033080): пациенты (n=398) в возрасте 12 лет и старше, у которых ген муковисцидозного трансмембранного регулятора проводимости (CFTR) характеризуется наличием одной мутации F508del и одной мутации с минимальной функциональностью (генотип F/MF);
SKYLINE 103 (NCT05076149): пациенты (n=573) в возрасте 12 лет и старше, у которых альтерации гена CFTR представлены следующим образом — либо две мутации F508del (генотип F/F), либо одна мутация F508del и гейтинг-мутация (генотип F/G), либо одна мутация F508del и мутация с остаточной функциональностью (генотип F/RF), либо отсутствие мутации F508del при наличии хотя бы одной мутации, чувствительной к тройной терапии муковисцидоза комбинированным препаратом «Трикафта» / «Кафтрио» (Trikafta / Kaftrio, элексакафтор + тезакафтор + ивакафтор, ивакафтор);
RIDGELINE 105 (NCT05422222): пациенты (n=78) в возрасте 6–11 лет с хотя бы одной CFTR-мутацией, чувствительной к назначению «Трикафта» / «Кафтрио».
Вначале во всех испытаниях участники прошли 4-недельный вводный период лечения при помощи «Трикафта» / «Кафтрио», после которого были оценены такие критические при муковисцидозе клинические показатели, как объем форсированного выдоха за 1-ю секунду (ОФВ1) в процентах от расчетного, уровень хлоридов в потовой жидкости и пр. Затем в первых двух исследованиях пациенты были рандомизированы на получение либо экспериментального «Алифтрек», либо стандартного «Трикафта» / «Кафтрио», в третьем (открытом) — только «Трикафта» / «Кафтрио». Результаты были сняты по прошествии 24 недель терапии.
В SKYLINE 102 и SKYLINE 103 эффективность комбинации из ванзакафтора, тезакафтора и деутивакафтора оказалась не хуже, чем результативность «Трикафта» / «Кафтрио», в том, что касается абсолютного изменения ОФВ1. Этот показатель, отражающий респираторную функцию легких, прибавил 0,5% и 0,2% — против его роста на 0,3% и 0,0% (p<0,0001) [1].
Назначение «Алифтрек» превзошло «Трикафта» / «Кафтрио» в задаче снижения концентрации хлоридов в потовой жидкости (ммоль/л), абсолютные изменения которой составили −7,5 и −5,1 — против +0,9 и −2,3 (p<0,0001 и p=0,0034), тем самым отразив клинически значимое улучшение канальной функции CFTR.
Объединенные данные таковы, что применение новой схемы лечения муковисцидоза в лице препарата «Алифтрек» вывело больше пациентов к концентрации хлоридов в потовой жидкости < 60 ммоль/л (то есть ниже диагностического порога) и < 30 ммоль/л (то есть ниже уровня у бессимптомных носителей). До первого порога добрались 86% испытуемых против 77% (p<0,0001) в группах «Трикафта» / «Кафтрио», до второго — 31% против 23% (p<0,0001).
Вероятность достижения первого порога при помощи экспериментальной терапии была приблизительно вдвое выше, второго порога — приблизительно втрое: соответствующие отношения шансов (odds ratio, OR) 2,21 (95% ДИ [здесь и далее]: 1,55–3,15) и 2,87 (2,00–4,12).
Клинические исходы RIDGELINE 105 продемонстрировали, что терапия муковисцидоза сочетанием ванзакафтора, тезакафтора и деутивакафтора позволила сохранить ОФВ1 на исходном уровне —изменение на 0,0% (−2,0, +1,9), — тогда как уровень хлоридов в потовой жидкости снизился на 8,6 ммоль/л (−11,0, −6,3). До пороговых концентраций < 60 ммоль/л и < 30 ммоль/л дошли 94,9% (87,4–98,6) и 52,6% (40,9–64,0) пациентов, притом что изначально их пропорции в этом статусе составляли 84% и 39%.
Каких-либо претензий к профилю безопасности «Алифтрек» не зафиксировано.
ЧТО ДАЛЬШЕ
Продолжается клиническое исследование NCT05422222 фазы III, тестирующее «Алифтрек» (Alyftrek, ванзакафтор + тезакафтор + деутивакафтор) в педиатрической популяции (1–11 лет) пациентов (n=210) с муковисцидозом и хотя бы одной CFTR-мутацией, чувствительной к назначению «Трикафта» / «Кафтрио» (Trikafta / Kaftrio, элексакафтор + тезакафтор + ивакафтор, ивакафтор).
Проводятся также клинические проверки долгосрочной безопасности, переносимости и эффективности «Алифтрек», NCT05444257 и NCT05844449 фазы III, — соответственно среди пациентов в возрасте 12 лет и старше и в возрасте 1 года и старше.
ЧТО ЕЩЕ
«Вертекс фармасьютикалс» (Vertex Pharmaceuticals) при поддержке «Модерна» (Moderna) работает над ингаляционным VX-522 (mRNA-3692) — экспериментальной мРНК-терапией муковисцидоза, предполагающей доставку в легкие генетических инструкций, кодирующих синтез нормального CFTR-белка. Лечение ориентировано на тех пациентов, у которых полностью отсутствует выработка CFTR, то есть модуляторы CFTR им не помогают. Продолжается соответствующее клиническое исследование NCT05668741 фазы I/II, первые результаты которого ожидаются к весне 2025 года.
ЭКСПЕРТНЫЕ КОММЕНТАРИИ
Появление «Трикафта» / «Кафтрио» (Trikafta / Kaftrio, элексакафтор + тезакафтор + ивакафтор, ивакафтор), первой в мире тройной противомуковисцидозной терапии, привело к резкому и беспрецедентному продлению жизни пациентов.
Согласно данным регистра пациентов Фонда муковисцидоза (Cystic Fibrosis Foundation, CFF, Бетесда, шт. Мэриленд, США), в 2022 году прогнозируемая медиана продолжительности жизни пациентов с муковисцидозом достигла 68,2 лет (95% ДИ: 63,0–76,2) — против 48,4 лет в 2019 году, который стал последним годом перед широкомасштабным внедрением «Трикафта» / «Кафтрио» в клиническую практику. Уровень смертности снизился с 1,2 летальных исходов на 100 человек в 2019 году — до 0,7 в 2022-м. Увеличение прогнозируемой продолжительности жизни на 20 лет сопровождалось улучшением состояния здоровья в отношении легочных и нелегочных проявлений муковисцидоза [1].
Новая тройная схема лечения муковисцидоза «Алифтрек» (Alyftrek, ванзакафтор + тезакафтор + деутивакафтор), которая пришла на смену «Трикафта» / «Кафтрио», обещает дополнительное улучшение состояния здоровья пациентов. Гипотеза прозрачна: непрерывное и устойчивое снижение концентрации хлоридов до значений, отмечаемых у бессимптомных носителей муковисцидоза, обеспечит должные благотворные эффекты, которые проявят себя на всем спектре поражаемых заболеванием органов и систем. Следует отметить, что некоторые положительные эффекты могут обнаруживаться только в долгосрочной перспективе при постоянном лечении.
Однократное ежедневное применение (вместо нынешних двух раз в день — утром и вечером) повысит приверженность лечению, особенно среди пациентов, принимающих несколько препаратов.
Тирзепатид, популярный препарат для похудения и гликемического контроля, стал первым лекарственным средством, разрешенным для лечения обструктивного апноэ во сне.
ОСНОВНЫЕ ФАКТЫ
С обструктивным апноэ во сне сталкивается великое множество людей во всём мире: 900 млн человек.
Основная рекомендация его лечения обращается к снижению лишнего веса как первопричины.
Поскольку агонисты рецептора глюкагоноподобного пептида-1 (GLP1RA) с прежде невиданной эффективностью способствуют избавлению от избыточной массы тела, их успех в лечении апноэ сна был предрешен. Но требовалась должная клиническая проверка.
Назначение тирзепатида (tirzepatide), за которым стоит «Илай Лилли» (Eli Lilly), на протяжении одного года действительно привело к тому, что у большинства пациентов заболевание перешло в легкую форму, а половина больных вообще избавилась от сонного апноэ.
В конце декабря 2024 года Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) одобрило применение тирзепатида в лечении умеренно-тяжелого обструктивного апноэ во сне у взрослых с ожирением [1].
В середине декабря 2024 года Европейское агентство по лекарственным средствам (EMA) высказалось одобрительно по вопросу использования тирзепатида в лечении обструктивного апноэ во сне, однако отказалось соответствующим образом расширять список его показаний: достаточно того, что этот GLP1RA-препарат назначается для снижения веса [2].
В России тирзепатид пока не зарегистрирован, но соответствующая клиническая проверка проводится; не исключено, он появится в виде недорогого дженерика.
ПРЯМАЯ РЕЧЬ
«Клиническая проверка показала, что люди с умеренно-тяжелым обструктивным апноэ во сне, получавшие тирзепатид, ежечасно испытывали приблизительно на 30 событий апноэ и гипопноэ меньше, причем половина пациентов вообще избавилась от данного патологического состояния. Если не лечить сонное апноэ, в долгосрочной перспективе это приведет к серьезным кардиометаболическим осложнениям».
Атул Малхотра (Atul Malhotra), директор по исследованиям в области пульмонологии, реаниматологии, медицины сна и физиологии в Медицинской школе Калифорнийского университета в Сан-Диего (UCSD, Сан-Диего, шт. Калифорния, США), бывший президент Американского торакального общества (ATS).
«От обструктивного апноэ во сне страдает огромное число людей, причем у многих оно выражено в умеренно-тяжелой форме, нарушающей повседневную жизнь. В 85% случаев это состояние остается недиагностированным, и, следовательно, не проходит лечения. В то время как существуют лекарственные препараты для лечения чрезмерной дневной сонливости по причине сонного апноэ, для непосредственного последнего фармакотерапии нет. Тирзепатид должен стать первым лекарством, причем эффективность лечения такова, что вспомогательную вентиляцию дыхательных путей можно отменить».
Джефф Эммик (Jeff Emmick), старший вице-президент по разработке продукции «Илай Лилли» (Eli Lilly).
СУТЬ ВОПРОСА
Обструктивное апноэ во сне характеризуется повторяющимся коллапсом глотки во время сна, приводящим к апноэ (остановка дыхательных движений) и гипопноэ (неполный вариант апноэ), с последующей гипоксемией (снижение кислорода в крови), гиперкапнией (повышение углекислого газа в крови) и повторяющимися пробуждениями [1].
Обструктивное апноэ во сне сопровождается клинически значимыми симптомами, такими как чрезмерная дневная сонливость, и является независимым фактором риска сердечно-сосудистых заболеваний [1] [2]. Это заболевание распространено и имеет значительные медицинские и экономические последствия. Свыше 900 млн человек в мире страдают от обструктивного апноэ во сне, и у 40% заболевание протекает в умеренной или тяжелой форме [3].
Лечение пациентов с обструктивным апноэ во сне исторически было сосредоточено на механической поддержке во время сна. Вентиляционная терапия положительным давлением в дыхательных путях (PAP) улучшает индекс апноэ–гипопноэ (AHI, число событий апноэ и гипопноэ в течение часа сна) и уменьшает симптомы, связанные с обструктивным апноэ во сне, но на общую эффективность лечения влияет различная приверженность терапии. Рандомизированные контролируемые испытания не продемонстрировали, что PAP-терапия снижает частоту неблагоприятных сердечно-сосудистых исходов и смертности [4] [5] [6].
Мандибулярная терапия (фиксирование нижней челюсти в выдвинутом положении специальным внутриротовым приспособлением) преимущественно используется для пациентов, которые не могут или не хотят следовать PAP-терапии, но она не является универсально эффективной [7]. Хирургия верхних дыхательных путей, включая стимуляцию подъязычного нерва, может быть эффективной, но, будучи инвазивным вмешательством, уместна лишь для отдельных пациентов.
Сейчас не существует одобренного фармакологического лечения обструктивного апноэ во сне.
Избыточная жировая масса является основным обратимым этиологическим фактором риска развития обструктивного апноэ во сне и его осложнений [8] [9]. Польза значительного снижения веса при лечении пациентов с обструктивным апноэ во сне хорошо известна, и клинические руководства настоятельно рекомендуют проводить лечение ожирения у таких пациентов [9]. Таким образом, фармакологическое вмешательство, направленное на борьбу с ожирением и его последующим влиянием на обструктивное апноэ во сне, симптомы, артериальное давление и системное воспаление низкого уровня, должно способствовать целостному подходу, который не может быть полностью достигнут при использовании вышеупомянутых механических методов терапии [10] [11] [12].
КЛИНИЧЕСКИЕ ПОДРОБНОСТИ
Клиническое испытание SURMOUNT-OSA (NCT05412004) фазы III (рандомизированное, двойное слепое, плацебо-контролируемое, многоцентровое, международное) изучило еженедельные подкожные инъекции тирзепатида (в максимально переносимой дозе 15 или 10 мг) среди взрослых пациентов (n=469) с умеренно-тяжелым обструктивным апноэ во сне (AHI ≥ 15 на полисомнографии) и ожирением (индекс массы тела [ИМТ] ≥ 30 кг/м2), которые либо не могли или не хотели обращаться к PAP-терапии (исследование #1),либо уже следовали PAP-терапии и планировали ей придерживаться на всём протяжении экспериментального лечения (исследование #2).
Среди исходных характеристик участников: мужчин 67% и 72% в исследовании #1 и исследовании #2 соответственно, в среднем 52 и 50 событий AHI в час, средний вес 115 и 116 кг, средний ИМТ 39,1 и 38,7 кг/м2, преддиабет у 65% и 57%.
По прошествии 52 недель лечения группы тирзепатида продемонстрировали статистически значимое (p<0,001) превосходство над группами плацебо, снизив AHI на 25 (95% ДИ [здесь и далее]: −29, −21) и 29 (−33, −25) события в час — против снижения на 5 (−9, −1) и 6 (−10, −1) события в час. Таким образом, разница с плацебо составила −20 (−26, −14) и −24 (−30, −18) события в час [1].
Если принимать во внимание данные только тех пациентов, которые полностью и строго прошли весь курс лечения, эффективность тирзепатида оказалась еще лучше: разница с плацебо составила −23 (−29, −16) и −24 (−30, −19) события в час.
У 61% и 72% испытуемых, получавших тирзепатид, число AHI-событий в час снизилось не менее чем наполовину, что является клинически значимым изменением, — против 19% и 23% в контрольных группах.
Статус ремиссии обструктивного апноэ во сне или его протекания в легкой бессимптомной форме был установлен для 42% и 50% участников в группах тирзепатида — против 16% и 14% в группах плацебо. Данный статус подтверждался выходом либо к числу AHI-событий в час < 5, либо к их количеству в пределах 5–14 и баллу ≤ 10 по Эпвортской шкале сонливости (ESS; диапазон от 0 до 24, больше — хуже), отражающей степень чрезмерной дневной сонливости.
Среди прочих показателей эффективности лечения синдрома обструктивного апноэ при помощи тирзепатида:
изменение гипоксической нагрузки, специфической для сонного апноэ (SASHB), % мин/ч: −95 (−103, −87) в исследовании #1 и −103 (−110, −96) в исследовании #2 — против −25 (−44, −6) и −42 (−64, −20) в группах плацебо;
изменение балла в пациентском опроснике PROMIS о нарушении сна (меньше — лучше): −6,6 (−8,2, −4,9) и −8,2 (−10,0, −6,3) — против −3,1 (−4,7, −1,6) и −3,9 (−5,9, −1,9);
изменение балла в пациентском опроснике PROMIS о расстройстве сна (меньше — лучше): −4,5 (−5,8, −3,1) и −7,0 (−8,6, −5,4) — против −2,4 (−3,8, −1,1) и −3,1 (−4,8, −1,4);
изменение массы тела, %: −18 (−19, −16) и −20 (−21, −18) — против −2 (−3, −0) и −2 (−4, −1);
изменение уровня высокочувствительного C-реактивного белка (hsCRP), мг/л: −1,4 (−1,7, −1,1) и −1,4 (−1,6, −1,1) — против −0,7 (−1,1, −0,3) и −0,3 (−0,8, +0,1);
изменение систолического артериального давления, мм рт. ст.: −10 (−12, −8) и −8 (−10, −6) — против −2 (−4, +0) и −4 (−6, −2);
изменение диастолического артериального давления, мм рт. ст: −5 (−6, −4) и −3 (−5, −2) — против −2 (−4, −1) и −2 (−4, −1).
Нежелательные явления (НЯ), возникавшие в ходе применения тирзепатида, в основном были связаны с желудочно-кишечным трактом (диарея, тошнота, рвота, запор), носили легко-умеренную степень тяжести, чаще возникали на этапе эскалации дозы.
Что касается серьезных НЯ, в группе тирзепатида в исследовании #2 были зарегистрированы два случая острого панкреатита. Случаев медуллярного рака щитовидной железы не зафиксировано. Пять участников (два в группах тирзепатида, три в группах плацебо) столкнулись с тяжелыми или серьезными депрессивными расстройствами, суицидальными мыслями или поведением.
ЭКСПЕРТНЫЕ КОММЕНТАРИИ
Назначение тирзепатида привело к тому, что число AHI-событий в час снизилось в среднем на 29, или на 59% относительно их исходного количества. Такое изменение считается клинически значимым: согласно Американской академии медицины сна, порог клинической значимости для данного показателя составляет 15 и более событий в час [1], тогда как, согласно другим источникам, к клинически значимым относится хотя бы 50-процентное снижение этого показателя [2] [3].
Учитывая, что значительная пропорция участников продемонстрировала статус ремиссии обструктивного апноэ во сне или его протекания в легкой форме, им можно было смело рекомендовать отказываться от PAP-терапии [4] [5] [6] [7].
Поскольку существенное снижение AHI-событий в час сопровождалось значимым улучшением гипоксического бремени, можно смело говорить об ожидаемом уменьшении риска сердечно-сосудистых осложнений и смерти [8] [9]. Что примечательно, данное снижение оказалось справедливым вне зависимости от использования или нет PAP-терапии.
Симптомы обструктивного апноэ во сне не только являются обременительными, но и несут за собой повышенный риск травматизма, в том числе связанный с автомобильными авариями и производственными травмами [10]. Тяжесть симптомов также служит предиктором повышенного риска сердечно-сосудистых осложнений [11]. Вот почему клинически значимым является факт улучшения качества сна, согласно опроснику PROMIS.
Сонное апное и ожирение — два разных, но тесно связанных заболевания, и оба они играют независимую этиологическую роль в развитии сердечно-сосудистых осложнений [12]. Согласно современным рекомендациям, пациентам с обструктивным апноэ во сне следует снизить вес хотя бы на 7–11% [13] [14], хотя недавний метаанализ установил, что оптимальным для дополнительного снижения AHI-событий в час является еще большее уменьшение массы тела [15]. Поскольку бариатрическая хирургия является инвазивной процедурой, вряд ли ее можно назвать целесообразной или подходящей для всех, а вот фармакотерапия тирзепатидом вполне себе подходит. Более того, тирзепатид принес дополнительную пользу в виде снижения артериального давления и воспаления.
Фармакологические усилия борьбы с болезнью Паркинсона путем избавления от нейротоксичного альфа-синуклеина выглядят не особо впечатляющими с точки зрения терапевтической эффективности — тем не менее игроки фармотрасли не сдаются.
СУТЬ ВОПРОСА
Болезнью Паркинсона, вторым после болезни Альцгеймера самым распространенным нейродегенеративным возрастным заболеванием, страдают свыше 10 млн человек в мире. К 2040 году число больных, как ожидается, достигнет 17 млн человек, приобретя форму пандемии [1] . Несмотря на многочисленные исследовательские усилия, болезнь Паркинсона по-прежнему остается неизлечимой [2].
Отличительными патологическими особенностями болезни Паркинсона являются дегенерация дофаминергических нейронов и отложения телец Леви, состоящих в основном из нерастворимых агрегатов, образованных неправильно свернутым альфа-синуклеином (синуклеопатия) — небольшим белком, состоящий из 140 аминокислот.
В последние годы гипотеза агрегации альфа-синуклеина привлекает большое внимание. Она постулирует, что в патологических условиях альфа-синуклеин подвергается мисфолдингу (неправильному сворачиванию), что приводит к его конформационным изменениям, вызывающим аномальную агрегацию. В ходе этого процесса образуются различные виды агрегатов альфа-синуклеина, включая олигомеры, протофибриллы и фибриллы, все из которых цитотоксичны для нейронов и способствуют их гибели. Особенно губительны промежуточные олигомеры и протофибриллы, причем растворимые протофибриллы обладают большей нейротоксичностью, чем нерастворимые зрелые фибриллы [3] [4].
Токсичные промежуточные продукты ответственны за запуск каскада событий, включая дисфункцию митохондрий, нейровоспаление, деформацию нейронов и ферроптоз, которые тесно связаны с патогенезом болезни Паркинсона. Они нарушают синаптическую передачу, функции органелл и целостность цитоскелета, структуру мембран и гематоэнцефалического барьера (ГЭБ) [5] [6].
Если говорить о митохондриях, которые являются основным источником реактивных форм кислорода (ROS) в клетках, то они особенно уязвимы для агрегатов альфа-синуклеина. Последние задействуют механизмы связывания с комплексами митохондриальной дыхательной цепи, взаимодействия с митохондриальными порами переходной проницаемости, вмешательства в импорт митохондриальных белков, нарушения контроля качества митохондрий [7].
Агрегация альфа-синуклеина нарушает работу аутофагического лизосомального пути (ALP) и убиквитин-протеасомной системы (UPS) — двух основных механизмов деградации неправильно свернутых и агрегированных белков, — что отражается изменениями внутриклеточного транспорта и снижением скорости клиренса поврежденных белков [8] [9].
В физиологических условиях микроглия и астроциты функционируют как соответственно ключевые иммунные и опорные клетки центральной нервной системы (ЦНС). Однако в присутствии патологического альфа-синуклеина пролиферация микроглии стимулируется такими механизмами, как активация толл-подобных рецепторов (TLR) и сигнальных путей p38/ATF2 и ядерного фактора κB (NF-κB). Всё это результирует выработкой различных факторов воспаления и ROS, способствуя дисфункции и гибели дофаминергических нейронов [10]. Альфа-синуклеиновые агрегаты активируют микроглию, которая дифференцируется в провоспалительный фенотип (фенотип M1) и вырабатывает провоспалительные факторы, хемокины и нейротоксические факторы, которые побуждают астроциты трансформироваться в нейротоксический фенотип (фенотип A1) [11], еще больше усиливающий активацию микроглии [12].
Альфа-синуклеиновые агрегаты распространяются по организму в первую очередь «прионоподобным» образом. Они также могут распространяться через туннелирующие нанотрубки (нанотуннели между клетками) [13], экзосомы [14] и посредством иных механизмов, что приводит к повсеместному отложению этих патологических белков по всему головному мозгу, особенно в неокортексе, гиппокампе, стриатуме, таламусе и мозжечке [6]. Аномальная агрегация альфа-синуклеина не ограничивается мозгом: она наблюдается в спинном мозге и периферической нервной системе, включая симпатические ганглии, блуждающий нерв, сердечные нервы и желудочно-кишечную систему [15].
Согласно гипотезе «кишечник — мозг», при болезни Паркинсона патологические процессы зарождаются в кишечнике и впоследствии распространяться на головной мозг [16]. Альфа-синуклеин вызывает изменения в составе микробиоты кишечника и кишечное воспаление, что приводит к дисфункции иммунной системы кишечника, энтеральной нервной системы и кишечного барьера. Активация глиальных клеток кишечника, повышенная кишечная проницаемость и окислительный стресс повышают уровень экспрессии альфа-синуклеина в кишечнике, что отражается мисфолдингом и аномальной агрегацией этого белка. Хроническое периферическое воспаление нарушает целостность ГЭБ, способствуя переносу альфа-синуклеина из кишечника в ЦНС [17] [18].
GLP1R-агонисты вроде ликсисенатида, эксенатида или семаглутида сдерживают прогрессирование моторных нарушений при болезни Паркинсона.
ПРАСИНЕЗУМАБ: PROTHENA / ROCHE
Ирландская «Проутина» (Prothena) совместно с «Рош» (Roche) доводит до ума гуманизированное моноклональное антитело прасинезумаб (prasinezumab, RG7935, PRX002, NEOD002) против агрегированного альфа-синуклеина. Но результаты пока не впечатляют.
Прасинезумаб, оригинатором которого является, «Неотоуп байосайенсиз» (Neotope Biosciences), связывается с человеческим агрегированным альфа-синуклеином с высоким сродством и авидностью. На мышиных моделях болезни Паркинсона и деменции с тельцами Леви прасинезумаб снижал уровень C-концевой усеченной формы альфа-синуклеина, которая считается нейротоксичной, а также сдерживал распространение альфа-синуклеина от клетки к клетке [1] [2] [3].
Клиническое исследование PASADENA (NCT03100149) фазы II, организованное среди пациентов (n=316) с недавно диагностированной болезнью Паркинсона (длительность заболевания не дольше 2 лет) и не придерживающихся заместительной дофаминой терапии, не смогло обеспечить выход к первичной конечной точке, заявленной улучшением общего балла по унифицированной рейтинговой шкале оценки болезни Паркинсона Международного общества изучения двигательных расстройств (MDS-UPDRS), которая оценивает тяжесть и прогрессирование этого заболевания [4] [5].
Однако затем выяснилось, что улучшения всё же есть, и они связаны с моторными функциями: годичный курс терапии из ежемесячных вливаний прасинезумаба привел к 35-процентному относительно плацебо сдерживанию прогрессирования моторных нарушений, такие как замедленность движений (брадикинезия), тремор, ригидность, нестабильность походки — согласно изменению балла части III шкалы MDS-UPDRS [6] [7]. При этом благотворный эффект замедления ухудшений, оказавшийся справедливым главным образом среди лиц с быстрым прогрессированием болезни Паркинсона, продолжал усиливаться в ходе продолжения лечения сроком максимум 4 года [8].
В мае 2021 года было запущено клиническое исследование PADOVA (NCT04777331) фазы IIb, проверяющее, насколько хорошо прасинезумаб сдерживает процесс ухудшения моторных функций у пациентов (n=586) с болезнью Паркинсона в более запущенной, чем в PASADENA, форме. Результаты будут раскрыты к концу 2024 года.
В середине декабря 2024 года стало известно о провале PADOVA: прасинезумаб не смог статистически значимым образом опередить плацебо, хотя численно (номинально) превзошел, на 16% отсрочив время до подтвержденного прогрессирования ухудшения двигательных функций — отношение риска (hazard ratio, HR) 0,84 (95% ДИ [здесь и далее]: 0,69–1,01; p=0,0657). В популяции пациентов, получавших терапию леводопой (75% испытуемых), эффект был более выраженным: HR 0,79 (0,63–0,99; p=0,0431). Отмечены устойчивые положительные тенденции по множеству вторичных и эксплоративных конечных точек [9] [10].
Согласно анализу данных, скорректированных по ряду ковариат (включая возраст, пол, стадию по Хён и Яру, применение фоновых медикаментов), терапевтический эффект прасинезумаба оказался номинально значимым: соответственно HR 0,81 (0,67–0,98; p=0,0334) и HR 0,76 (0,61–0,95; p=0,0175).
МИНЗАСОЛМИН И UCB7853: UCB / NOVARTIS
В декабре 2021 года бельгийская «ЮСиБи» (UCB) договорились с «Новартис» (Novartis) о совместной разработке и коммерциализации двух лекарственных активов, предназначенных для лечения болезни Паркинсона [1].
По условиям соглашения,«ЮСиБи» получит от «Новартис» авансом $150 млн и последующие выплаты на сумму до $1,5 млрд долларов по мере прохождения определенных этапов развития экспериментальных проектов. В случае коммерциализации готовых лекарств доходы будут делиться территориально: «ЮСиБи» займется их реализацией в Европе и Японии, «Новартис» — в США и других странах.
Партнерство касается двух препаратов-кандидатов, нацеленных на альфа-синуклеин.
Минзасолмин (minzasolmin, UCB0599,NPT200-11, DLX-313), который «ЮСиБи» в 2014 году лицензировала у «Ньюропор терапис» (Neuropore Therapies), ингибирует внутриклеточную агрегацию альфа-синуклеина. Низкомолекулярный минзасолмин, будучи циклической пептидомиметической молекулой, является соединением второго поколения, оптимизированным для пероральной биодоступности и прохождения через гематоэнцефалический барьер.
Минзасолмин, который взаимодействует с C-концевым доменом альфа-синуклеина и предотвращает его связывание с мембранами с дальнейшей олигомеризацией в них, in vitro сдерживает агрегацию альфа-синуклеина, а на трансгенных мышиных моделях нормализует нейронные и воспалительные маркеры, устраняет моторный дефицит, ослабляет кортикальную альфа-синуклеиновую патологию и астроглиоз, нормализует уровень стриарного дофаминового транспортера [2] [3].
В декабре 2020 года «ЮСиБи» приступила к 18-месячному клиническому исследованию ORCHESTRA (NCT04658186) фазы IIa (рандомизированному, двойному слепому, плацебо-контролируемому, многоцентровому, международному), проверяющему терапевтическую эффективность минзасолмина среди пациентов (n=496) с болезнью Паркинсона на ранней стадии (стадия паркинсонизма ≤ 2,5 по Хён и Яру, длительность заболевания не дольше 2 лет). Результаты должны быть собраны ближе к концу 2024 года.
В середине декабря 2024 года стало известно о провале ORCHESTRA: минзасолмин не смог обеспечить выход ни к первичной конечной точке эффективности лечения болезни Паркинсона (изменение суммарного балла частей I–III шкалы MDS-UPDRS), ни к вторичным (изменения баллов отдельных частей этой шкалы) — экспериментальное лечение не замедлило прогрессирование заболевания [4].
Продолжается клиническое испытание NCT05543252 фазы II, изучающее долгосрочные безопасность, переносимость и эффективность минзасолмина при впервые поставленном диагнозе болезни Паркинсона пациентам (n=428) из других исследований этого экспериментального препарата. Оно завершится в 2027 году.
Отсутствие доказательств клинической пользы минзасолмина привело к прекращению NCT05543252.
UCB7853 — моноклональное антитело против альфа-синуклеина, подавляющее его внеклеточное распространение.
В декабре 2020 года было запущено клиническое исследование NCT04651153 фазы I, оценивающее различные внутривенные дозы UCB7853 с позиций безопасности, переносимости и фармакокинетики — среди здоровых добровольцев и пациентов с болезнью Паркинсона на легко-умеренной стадии. Испытание завершено, дальнейших шагов по развитию проекта пока не предпринималось.
В середине декабря 2024 года, после клинической неудачи минзасолмина, «ЮСиБи» подтвердила свою приверженность продолжению разработки UCB7853 [4].
В свое время утверждалось, что UCB0599 и UCB7853 органично дополняют друг друга в задаче замедления прогрессирования болезни Паркинсона: первый препарат показан на ранних ее стадиях, второй подходит при более запущенных состояниях.
Дофаминомиметик тавападон предназначен для улучшения моторных функций при паркинсонизме без обременяющих побочных эффектов.
ЭКСИДАВНЕМАБ: BIOARCTIC / ABBVIE
В руках «ЭббВи» (AbbVie) было гуманизированное моноклональное антитело эксидавнемаб (exidavnemab, ABBV-0805, BAN0805), в декабре 2018 года лицензированное у шведской «Байоарктик» (BioArctic) и связывающее олигомерные и протофибриллярные формы альфа-синуклеина различных конформаций с пикомолярной аффинностью [1] [2].
Эксидавнемаб характеризуется очень высокой (100 тыс. раз) селективностью связывания агрегированного альфа-синуклеина относительно его мономерных форм. Для сравнения: сродство прасинезумаба (prasinezumab, RG7935, PRX002) и цинпанемаба (cinpanemab, BIIB054), за которыми стоят «Рош» (Roche) / «Проутина» (Prothena) и «Байоджен» (Biogen) / «Ньюримьюн» (Neurimmune), к агрегированному альфа-синуклеину соответственно в 400 и 800 раз выше, чем к мономерному.
Учитывая высокий уровень физиологических мономеров альфа-синуклеина в крови, важно минимизировать его связывание, во-первых, во избежание периферической секвестрации антитела в плазме, чтобы его большее количество достигло своих мишеней в головном мозге, и, во-вторых, для улучшенного профиля безопасности и сниженной терапевтической дозы.
Согласно клинической проверке среди здоровых добровольцев (n=95), эксидавнемаб, назначаемый однократно внутривенно по 300–6000 мг или подкожно по 300 мг, сохраняет свою активность продолжительное время: период полувыведения составляет приблизительно 30 дней. Установлено дозозависимое снижение плазматической концентрации свободного альфа-синуклеина. Каких-либо проблем с безопасностью не выявлено [3].
Будущее эксидавнемаба казалось туманным, поскольку в июле 2020 года «ЭббВи» по стратегическим причинам остановила соответствующее клиническое испытание NCT04127695 фазы I. Впоследствии компания вышла из партнерства с «Байоарктик», которая стала самостоятельно развивать этот препарат-кандидат [4].
В ноябре 2024 года было организовано клиническое исследование EXIST (NCT06671938) фазы IIa среди пациентов (n=24) с легко-умеренной болезнью Паркинсона, следующих стабильным курсом симптоматической терапии. Завершение запланировано на 2026 год. По результатам «Байоарктик» примет окончательное решение, что делать с эксидавнемабом: сворачивать проект либо продолжать им заниматься в целях лечения болезни Паркинсона, деменции болезни Паркинсона, деменции с тельцами Леви или множественной системной атрофии.
PD01A: AC IMMUNE
Несмотря на скудные успехи игроков «Большой фармы», швейцарская «ЭйСи имьюн» (AC Immune) верит в разумность гипотезы альфа-синуклеина: для этого в июле 2021 года у австрийской «Аффирис» (AFFiRiS) была куплена PD01A — активная вакцина против альфа-синуклеина [1].
Иммуноген в составе PD01A представляет собой пептид из восьми аминокислот (PD01), который имитирует эпитоп в C-концевой области человеческого альфа-синуклеина, но с другой аминокислотной последовательностью. Пептид конъюгирован с белком-носителем гемоцианином лимфы улитки (KLH) и адсорбирован на адъювантном гидроксиде алюминия. Вакцина PD01A разработана таким образом, чтобы стимулировать В-клеточный антительный ответ, но обходить аутореактивную мобилизацию Т-клеток, которая может вызвать ненужные нейровоспалительные реакции.
Активная иммунизация при помощи PD01A генерирует антитела, которые избирательно распознают агрегаты альфа-синуклеина с гораздо меньшим сродством к его мономерным формам и без реактивности к бета-синуклеину. Белок-носитель обеспечивает необходимые эпитопы Т-хелперов для индукции длительного и усиленного ответа антител, тогда как антигенный компонент (PD01) действует исключительно как В-клеточный эпитоп и отвечает за специфичность гуморального иммунного ответа [2].
В июле 2023 года «ЭйСи» запустила клиническое испытание VacSYn (NCT06015841) фазы II, которое изучает экспериментальную вакцину ACI-7104.056, представляющую собой PD01 в оптимизированной рецептуре, среди пациентов (n=150) с болезнью Паркинсона на начальной стадии. К концу 2024 года будет предоставлен первый промежуточный отчет о безопасности и иммуногенности вакцины. Срок окончания исследования — начало 2028 года.
Ничего не получилось у «Байоджен» (Biogen) с моноклональным антителом цинпанемаб (cinpanemab, BIIB054), лицензированным у швейцарской «Ньюримьюн» (Neurimmune) и таргетированным против агрегатов альфа-синуклеина. Хотя в отрасли считалось, что этот препарат наиболее совершенный с технической точки зрения. В клиническом испытании SPARK (NCT03318523) фазы II цинпанемаб не обеспечил каких-либо значимых улучшений по шкале MDS-UPDRS. В феврале 2021 года дальнейшая разработка цинпанемаба была прекращена [1].
ABL301: ABL BIO / SANOFI
В середине января 2022 года «Санофи» (Sanofi) приобрела у корейской «Эй-би-эл байо» (ABL Bio) мировую лицензию на экспериментальный препарат ABL301 (SAR446159), предназначенный для лечения болезни Паркинсона [1].
Сделка предполагает выплату $75 млн авансом и последующей суммы до $985 млн по мере развития проекта, а также роялти от продаж готового лекарства.
ABL301 представляет собой биспецифическое моноклональное антитело, таргетированное на альфа-синуклеин и рецептор инсулиноподобного фактора роста 1 (IGF1R) [2] [3].
ABL301 с высокой аффинностью распознаёт патологические агрегаты альфа-синуклеина, обходя стороной его мономерные формы. Нацеливание на IGF1R, сделанное в рамках технологической платформы Grabody-B, позволяет ABL301 без проблем миновать гематоэнцефалический барьер (ГЭБ). Поскольку IGF1R высоко и относительно специфично экспрессирует на эндотелиальных клетках головного мозга, ABL301 проникает в мозг посредством рецептор-опосредованного трансцитоза (RMT).
Как утверждает «Эй-би-эл», ее ABL301 характеризуется рядом выгодных преимуществ перед другими моноклональными антителами, которые разрабатывают игроки фармотрасли. Во-первых, биологические лекарственные препараты конкурентов весьма плохо проникают через ГЭБ: в головном мозге обнаруживается приблизительно 0,1–0,2% таких антител.
Во-вторых, биопрепараты соперников фактически без особого разбора связывают агрегированные и мономерные формы альфа-синуклеина, хотя именно первые вовлечены в патофизиологические каскады при болезни Паркинсона.
В-третьих, ABL301 располагает двойным механизмом действия: содействие фагоцитозу внеклеточного альфа-синуклеина микроглиальными клетками и подавление прионоподобного распространения альфа-синуклеина от клетки к клетке. Антитела конкурентов в основном осуществляют только второе.
Продолжается клиническое испытание NCT05756920 фазы I среди здоровых добровольцев (n=86). Его завершение намечено к началу 2025 года.
БУНТАНЕТАП: ANNOVIS BIO
Интересным видится подход «Анновис байо» (Annovis Bio): ее низкомолекулярный препарат-кандидат бунтанетап (buntanetap, ANVS401), также известный как посифен (posiphen), нацелен сразу на три нейротоксичных белка — бета-амилоид, альфа-синуклеин и тау-белок. Все они нарушают аксональный транспорт нейромедиаторов и нейротрофических факторов и замедляют синаптическую передачу, тем самым ухудшая нервную деятельность в целом. Подобные нарушения приводят к активации иммунной системы, которая атакует нервные клетки, что приводит к нейровоспалению, дегенерации и смерти нервных клеток. Итогом становится ухудшение когнитивной и моторной деятельности.
Клиническое исследование NCT04208152 фазы I протестировано эмрусолмин среди взрослых добровольцев (n=68), установив его приемлемые безопасность и переносимость [1].
Клиническое испытание NCT04685265 фазы Ib проверило назначение различных доз эмрусолмина пациентам с легко-умеренной болезнью Паркинсона. Исследование, завершившееся в конце 2022 года, результатов так и не предоставило.
В конце октября 2021 года к проекту эмрусолмина подключилась «Тева фармасьютикал индастриз» (Teva Pharmaceutical Industries), пожелавшая обзавестись мировыми правами на этот препарат-кандидат [2]. Партнерство также касается sery433 — пролекарства эмрусолмина, разработанного с прицелом повышения его биодоступности [3].
«Тева» осуществляет 56-недельное клиническое испытание TOPAS-MSA (NCT06568237) фазы II, изучающее эмрусолмин в лечении множественной системной атрофии. Результаты будут собраны к середине 2027 года.
Пероральный низкомолекулярный эмрусолмин, эффективно преодолевающий гематоэнцефалический барьер (ГЭБ), специфически и с наномолярной аффинностью связывает токсичные олигомерные структуры внутриклеточного альфа-синуклеина, что приводит к их растворению и предотвращению образования новых олигомеров.
Поскольку эмрусолмин разрушает преамилоидные олигомеры и нарушает рост фибрилл, это приводит, в сочетании с механизмами клеточного клиренса, к уменьшению количества отложенных амилоидных фибрилл. Эмрусолмин не является разрушителем фибрилл, что важно, так как их фрагментация усиливает прионоподобное распространение токсичных белков и прогрессирование болезни за счет образования большего количества частиц, способных привлекать мономеры путем неправильного фолдинга.
Эмрусолмин, будучи дифенилпиразольным соединением, подавляет образование олигомеров альфа-синуклеина и прионного белка. На различных мышиных моделях синуклеопатий и болезни Паркинсона эмрусолмин препятствовал накоплению олигомеров и дегенерации нейронов [4], улучшил функцию дофаминовых нейронов и моторные функции [5], сдержал прогрессирование заболевания, причем даже после клинической манифестации болезни Паркинсона [6].
Утверждается также, что поскольку патологические олигомеры при нейродегенеративных заболеваниях имеют общие структурные особенности, хотя основной белковый компонент специфичен для каждой болезни, а эмрусолмин модулирует образование олигомеров путем воздействия на структурно-зависимые эпитопы, эта молекула, не исключено, может найти себя в лечении различных патологий, связанных с агрегацией белков [4].
На мышиных моделях множественной системной атрофии эмрусолмин снизил количество олигомеров альфа-синуклеина, сохранил дофаминергические нейроны и улучшил ходьбу [7], сдержал нейродегенерацию в черном веществе и уменьшил микроглиальную активацию [8]. На модели, имитирующей более тяжелой форму этого заболевания, эмрусолмин несколько улучшил двигательные навыки, но существенно не повлиял на нейродегенерацию или накопление альфа-синуклеина [9].
На мышиных моделях тау-патологии эмрусолмин уменьшил потерю нейронов, улучшил когнитивные способности и продлил выживаемость [10], причем даже у старых мышей он смог обратить вспять нарушения конечного метаболизма глюкозы в головном мозге [11].
На мышиной модели болезни Альцгеймера эмрусолмин восстановил синаптическую пластичность гиппокампа и память [12].
АМБРОКСОЛ
Амброксол (ambroxol) —муколитическое лекарственное средство и основной ингредиент ряда безрецептурных противокашлевых препаратов. В США и Канаде амброксол не зарегистрирован.
Интерес к изучению амброксола в лечении болезни Паркинсона обусловлен его активностью в качестве молекулярного шаперона для лизосомального фермента бета-глюкоцереброзидазы (GCase): мутации с потерей функции в гене GCase (GBA1) ассоциированы с повышенным в 20–30 раз риском развития болезни Паркинсона, причем с более ранним началом и ускоренными когнитивными и моторными ухудшениями [1] [2].
Активность бета-глюкоцереброзидазы и альфа-синуклеина связаны между собой: дефицит GCase вызывает патологическое накопление альфа-синуклеина в клеточных культурах [3], а сверхэкспрессия GCase в головном мозге ослабляет патологию и дефицит памяти в мышиной модели синуклеинопатии [4]. Активность GCase снижена при идиопатической болезни Паркинсона без мутаций GBA1 [5], снижение активности GCase коррелирует с более ранним началом заболевания и ухудшением когнитивных и немоторных симптомов [6]. Мутации GBA1 были первоначально идентифицированы [7] как причина [8] паркинсонизма при болезни Гоше — лизосомальном заболевании, характеризующемся накоплением сфинголипидов и в некоторых случаях патологией альфа-синуклеина.
В клетках, полученных от пациентов с болезнью Гоше или болезнью Паркинсона, амброксол стабилизировал мутантную GCase [9] и способствовал ее перемещению из эндоплазматического ретикулума (ER) в лизосомы [10], повышая уровень белка, активность фермента и функцию лизосом. У мух с мутациями GCH1 амброксол усиливал активность GCase, снижал стресс ER и защищал двигательную функцию [11].
У мышей дикого типа и мышей с мутацией GCH1 амброксол индуцировал активность GCase в головном мозге, а у мышей, сверхэкспрессирующих человеческий альфа-синуклеин, снижал общее количество и уровень фосфорилированного альфа-синуклеина [12]. В мышиной модели бокового амиотрофического склероза амброксол улучшал двигательную функцию и продлевал выживаемость [13].
У здоровых нечеловекообразных приматов ежедневный прием амброксола усиливал активность GCase в головном мозге [14].
Клиническое испытание AiM-PD (NCT02941822) фазы II, проведенное Университетским колледжем Лондона (University College London, UCL, Лондон, Великобритания), продемонстрировало, что ежедневные 1260 мг амброксола (многократно увеличенная доза, применяемая при кашле), назначаемые в течение пяти месяцев, способствовали улучшению показателей по шкале MDS-UPDRS (в особенности моторных функций) у пациентов с болезнью Паркинсона умеренной тяжести [15].
Лоусоновский научно-исследовательский институт здоровья (Lawson Health Research Institute, London, Онтарио, Канада) осуществляет клиническую проверку NCT02914366 фазы II на предмет того, сможет ли амброксол улучшить когнитивные и моторные симптомы при деменции на фоне болезни Паркинсона. Сроки завершения установлены на конец 2025 года [16].
Анализ реальных данных пациентов с болезнь Гоше, в том числе страдающих болезнью Паркинсона на ее фоне, которые принимали амброксол, подтвердил оказываемые им благотворные эффекты, проявляющиеся стабилизацией или улучшением неврологического статуса, увеличением физической активности, снижением утомляемости [17].
ВЕКТОРИЗИРОВАННЫЕ АНТИТЕЛА: VOYAGER THERAPEUTICS
Интересную идею продвигает «Вояджер терапьютикс» (Voyager Therapeutics), придумавшая решение проблемы доставки в головной мозг необходимого количества антител, связывающих патологические белки, в том числе альфа-синуклеин. «Вояджер» занялась вопросом обхождения гематоэнцефалического барьера (ГЭБ), препятствующего проникновению крупных лекарственных молекул. Да, можно осуществлять частые системные инъекции антител, но это не выход, поскольку высока вероятность побочных реакций.
Принцип так называемых векторизированных антител следующий: конструируется аденоассоциированный вирусный вектор (AAV), который несет определенные генетические инструкции, кодирующие нужное моноклональное антитело, синтез которого осуществляется самим организмом прямиком в головном мозге. Заявлено, что однократной внутривенной инъекции AAV-препарата окажется достаточно для оказания должного терапевтического эффекта очень продолжительное время — возможно, пожизненно. Объясняется это тем, что клетками-мишенями в центральной нервной системы в данном случае являются нейроны, долгоживущие и неделящиеся [1].
Всё бы ничего, но в августе 2020 года «ЭббВи» (AbbVie)разорвала с «Вояджер» мощное партнерское соглашение по векторизированным антителам против нейродегенеративных заболеваний.
ПРИРОДНЫЕ ВЕЩЕСТВА
Природные вещества (ПВ) — это соединения, полученные из природных источников, таких как растения, животные и микроорганизмы. С развитием современной химии и фармакологии выделение и характеристика ПВ становятся всё более сложными, открывая новые возможности для поиска лекарств. Многие ПВ обладают значительной биологической активностью и проявляют многообещающие терапевтические эффекты при широком спектре заболеваний.
Определенные природные вещества обладают способностью подавлять агрегацию альфа-синуклеина [1], однако клинических исследований, которые бы безговорочно подтвердили это чрезвычайно мало. Поэтому приходится полагаться либо на теоретические изыскания, либо на ограниченные данные применения ПВ при болезни Паркинсона.
Большую группу среди таких противопаркинсонических ПВ занимают полифенольные соединения: куркумин (curcumin) и его аналоги и производные, ресвератрол (resveratrol), пицеатаннол (piceatannol), катехины зеленого чая, гесперетин (hesperetin) и гесперидин (hesperidin), дигидромирицетин (dihydromyricetin, DHM) и сальвианоловая кислота B (salvianolic acid B, Sal B), рутин (rutin), текторигенин (tectorigenin), байкалеин (baicalein).
Способностью противодействовать нейротоксичным альфа-синуклеиновым агрегатам обладают следующие природные вещества: нафтохиноны (шиконин [shikonin], витамин K), таншиноны, экстракты грибов Ganoderma spp. («Линчжи», «Рейши», «Гриб бессмертия»), экстракты центеллы азиатской (Centella asiatica), производные коричной кислоты (cinnamic acid), экстракты гравилата городского (Geum urbanum), (+)-десдиметилпинорезинол [(+)-desdimethylpinoresinol], алкалоиды (хинолины и индолы, никотин, кофеин, скваламин [squalamine], тродускумин [trodusquemine]), а также пирролохинолинхинон (pyrroloquinoline quinone).
«Имфинзи» (Imfinzi, дурвалумаб) стал первым лекарственным препаратом, разрешенным в лечении мелкоклеточного рака легкого (МРЛ) на локализованной стадии, который не прогрессировал после одновременного проведения платиносодержащей химиотерапии и лучевой терапии.
ОСНОВНЫЕ ФАКТЫ
Соответствующий регуляторный вердикт вынесен Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) в начале декабря 2024 года [1].
«АстраЗенека» (AstraZeneca) доказала уместность длительного 24-месячного назначения дурвалумаба (durvalumab), блокатора PD-L1, в консолидационных целях, то есть для предупреждения прогрессирования или рецидива МРЛ на стадии I–III.
Применение «Имфинзи» существенно продлило важнейшие для пациента клинические показатели — общую выживаемость и выживаемость без прогрессирования.
Подключение иммунотерапевтического дурвалумаба к лечению МРЛ — еще один шаг к победе над этим агрессивным онкологическим заболеванием, оставляющим в живых не более трети пациентов в течение пяти лет после постановки диагноза.
«Имфинзи» уже используется в первоочередном лечении МРЛ на запущенной (распространенной) стадии.
СУТЬ ВОПРОСА
Мелкоклеточный рак легкого (МРЛ) — агрессивный вид рака, на который приходится приблизительно 15% случаев всех опухолей легких [1]. У трети пациентов с диагнозом МРЛ наблюдается локализованная (ограниченная) стадия заболевания [2], при которой наибольшая выживаемость достигается при одновременном проведении торакальной химиорадиотерапии с использованием этопозида и платиносодержащих препаратов (цисплатина или карбоплатина) и ранней торакальной радиотерапии с последующим профилактическим облучением черепа, если это показано [1] [3] [4].
Однако у большинства пациентов рецидив заболевания возникает в течение 2 лет после начала лечения [5] [6], а общая выживаемость в течение 5 лет не превышает 29–34% [7] [8] [9] [10] [11].
За последние три десятилетия не было достигнуто никаких успехов в системном лечении локализованного МРЛ [2] [12].
Некоторое улучшение выживаемости пациентов с мелкоклеточным раком легкого на локализованной стадии произошло после внедрения одновременной химиорадиотерапии на основе платины и ранней торакальной радиотерапии у пациентов, которые были достаточно здоровы для проведения подобного лечения [13]. И всё же плохие результаты в отдаленной перспективе вынудили искать альтернативные схемы радиотерапии и системной терапии. Торакальная радиотерапия дважды в день вначале казалась передовым методом лечения [14], но метаанализ показал, что общая выживаемость и частота токсических эффектов аналогичны тем, которые регистрировались при радиотерапии один раз в день [15]. Ряд исследований адъювантной и поддерживающей системной терапии также не продемонстрировали значительного улучшения результатов: бевацизумаб (bevacizumab), ниволумаб (nivolumab) с ипилимумабом (ipilimumab), интерферон альфа-2a (interferon alfa-2a), бацилла Кальметта — Герена (БЦЖ) и другая фармакотерапия — ничего не помогло улучшить исходы [16] [17] [18] [19].
КЛИНИЧЕСКИЕ ПОДРОБНОСТИ
Клиническое исследование ADRIATIC (NCT03703297) фазы III (рандомизированное, двойное слепое, плацебо-контролируемое, многоцентровое, международное) пригласило взрослых пациентов (n=) с мелкоклеточным раком легкого на локализованной стадии (неоперабельная стадия I–II, стадия III), заболевание которых не прогрессировало после радикальной химиорадиотерапии [1].
Среди исходных характеристик участников:
медиана возраста: 62 года;
бывших табакокурильщиков: 69%;
заболевание на стадии III: у 87%;
химиорадиотерапию цисплатином с этопозидом прошли 66%, карбоплатином с этопозидом — 34%;
облучение один раз в день прошли 72%, два раза в день — 28%.
Испытуемым назначали либо плацебо, либо дурвалумаб, либо дурвалумаб с тремелимумабом — на протяжении максимум 24 месяцев (в этот период отмечается наибольший риск рецидива), до момента прогрессирования заболевания или неприемлемой токсичности. Впрочем, позже от тремелимумаба полностью отказались.
После наблюдений в течении медианных 37,2 месяца (0,1–60,9) оценочная медиана общей выживаемости (OS) в группе дурвалумаба составила 55,9 месяца (95% ДИ [здесь и далее]: 37,3–NE) — против 33,4 месяца (25,5–39,9) в группе плацебо. Таким образом, назначение дурвалумаба привело к снижению риска смерти на относительных 27%: отношение риска (hazard ratio, HR) 0,73 (98,3% ДИ: 0,54–0,98; p=0,01) [2].
Вероятность остаться в живых на протяжении 24 и 36 месяцев получилась равной 68% и 57% среди получавших «Имфинзи» — против 59% и 48% в контрольной группе.
Медиана выживаемости без прогрессирования (PFS) вышла к 16,6 месяца (10,2–28,2) — против 9,2 месяца (7,4–12,9), то есть риск прогрессирования заболевания или смерти снизился на относительных 24%: HR 0,76 (97,2% ДИ: 0,59–0,98; p=0,02).
18- и 24- месячные частоты PFS определились на уровне 49% и 46% — против 36% и 34%.
Частота общего ответа (ORR) среди тех, у кого заболевания поддавалось количественной оценке, составила 30% (24–38), включая 3% полных ответов (CR) и 27% частичных ответов (PR), — против 32% (25–40), в том числе CR 2% и PR 30%.
Медиана длительности ответа (DOR) зафиксировалась на 33,0 месяца (22,4–NR)— против 27,7 месяца (9,6–NR). Ответ на протяжении 12 и 18 месяцев сохранялся у 74% (59–84) и 71% пациентов (57–82) — против 60% (44–73) и 55% (39–68).
ЭКСПЕРТНЫЕ КОММЕНТАРИИ
Клиническое испытание ADRIATIC (NCT03703297) фазы III подтвердило, что применение адъювантной терапии дурвалумабом после радикальной химиорадиотерапии мелкоклеточного рака легкого (МРЛ) на локализованной стадии привело к значительному улучшению, если сравнивать с плацебо, общей выживаемости (OS) и выживаемости без прогрессирования (PFS).
При стандартной химиолучевой терапии медиана OS составляет 25–30 месяцев, а 5-летняя OS — 29–34% [1] [2] [3] [4] [5]. Согласно результатам ADRIATIC, OS на протяжении 3 лет вышла к 57%.
Любопытно, в группе плацебо медиана OS и 3-летняя OS получились равными 33,4 месяца и 48%, что превышает показатели предыдущих исследований фазы III [1] [2] [3]. К примеру, в CONVERT (NCT00433563) медиана OS получилась равной 25,4–30,0 [2] [3].
Это, возможно, объясняется тем, что, во-первых, в ADRIATIC были включены только такие пациенты, у которых не было прогрессирования МРЛ после химиорадиотерапии, и исключены те, у кого сохранялись токсические эффекты умеренной и худшей степени выраженности; во-вторых, методы химиолучевой терапии усовершенствовались [1] [6]; в-третьих, появился доступ к схемам химиоиммунотерапии для пациентов, у которых развился отдаленный рецидив [7].
Тем не менее результаты ADRIATIC примечательны, особенно если учитывать, что у большинства пациентов с МРЛ на локализованной стадии рецидив или прогрессирование заболевания происходит в течение 2 лет после начала лечения [1] [2] [3] [4] [5] [8] [9] [10].
Поскольку адъювантный дурвалумаб продлил PFS, а 71% пациентов, его получавшие, демонстрировали ответ на лечение даже после 18 месяцев терапии (против 55% в группе плацебо), имеет смысл придерживаться длительного 24-месячного назначения дурвалумаба, ведь именно в этом периоде больные подвержены наибольшему риску рецидива.
Результаты ADRIATIC согласуются с данными, собранными в PACIFIC (NCT02125461) фазы III, в котором пациенты с местнораспространенным неоперабельным мелкоклеточным раком легкого (НМРЛ) без прогрессирования после химиолучевой терапии извлекли преимущество в OS [11] и PFS [12], если пользовались адъювантным дурвалумабом [13].
Можно предположить, что химиолучевая терапия оказывает обширное действие, основанное на механизме, при помощи которого микроокружение опухоли легкого становится более чувствительным к последующей иммунотерапии, как это было описано при других опухолях [14]. И потому необходимы дальнейшие исследования биологического влияния химиорадиотерапии на следующую за ней иммунотерапию, в том числе при различных подтипах МРЛ [15], с соответствующим изучением потенциальных биомаркеров.
Продолжается оценка других иммунотерапевтических стратегий в контексте МРЛ на локализованной стадии, как то: PD-L1-блокатор сугемалимаб (sugemalimab), DLL3-таргетированный тарлатамаб (tarlatamab), PD-L1-блокатор атезолизумаб (atezolizumab), PD-1-блокатор пембролизумаб (pembrolizumab) с опциональным PARP-ингибитором олапарибом (olaparib), PD-L1-блокатор адебрелимаб (adebrelimab) [16] [17] [18] [19] [20].
Расширенное применение иммунотерапии, обозначенное в ADRIATIC, повлияет на общую парадигму лечения мелкоклеточного рака легкого на локализованной стадии: ранняя (считай, предупредительная, консолидационная) иммунотерапия отразится на эффективности последующего ее назначения в случае прогрессирования заболевания. Как именно — это еще предстоит выяснить.
Болезнь Паркинсона — распространенное и изнуряющее нейродегенеративное заболевание.
Никакое из лекарств не в силах остановить неуклонное прогрессирование болезни Паркинсона, сопровождающееся нарастающей инвалидизацией.
GLP1R-агонисты, вовсю применяемые в лечении сахарного диабета и ожирения, открылись с неожиданной стороны.
Собраны многочисленные доказательства, что препараты вроде семаглутида обладают нейропротекторным действием, сдерживающим ухудшение двигательных функций при болезни Паркинсона.
Параллельно GLP1R-агонисты изучаются в лечении неалкогольного стеатогепатита (НАСГ), болезни Альцгеймера, инсульта, алкоголизма.
ЧТО ПРОИЗОШЛО
Добавление препарата «Адликсин» / «Ликсумия» (Adlyxin / Lyxumia, ликсисенатид) к стандартной терапии болезни Паркинсона привело к замедлению процесса ухудшения двигательных (моторных) функций, таких как дрожание конечностей (тремор), медлительность и скованность движений, трудности с удержанием равновесия.
Ликсисенатид (lixisenatide) относится к классу агонистов рецептора глюкогоноподобного пептида 1 (GLP1R) — такому же, к которому принадлежат мегапопулярные «Оземпик» (Ozempic, семаглутид), «Вегови» (Wegovy, семаглутид), «Мунджаро» (Mounjaro, тирзепатид) и «Зепбаунд» (Zepbound, тирзепатид), разработанные «Ново Нордиск» (Novo Nordisk) и «Илай Лилли» (Eli Lilly) и вовсю применяемые в лечении сахарного диабета 2-го типа и ожирения.
В России семаглутид, защищенный патентами оригинатора до 2035 года, но в конце 2023 года получивший принудительную лицензию на производство, доступен в виде следующих препаратов: «Семавик» (Semavic), «Квинсента» (Queensenta) и «Инсудайв» (Insudive). Эти генерические лекарственные средства никак и ничем не уступают брендовым препаратам, притом что стоят существенно дешевле. Тирзепатид пока не зарегистрирован, но соответствующая клиническая проверка проводится; не исключено, он появится в виде недорогого дженерика.
«Адликсин» / «Ликсумия», в свое время продвигавшийся «Санофи» (Sanofi), более не реализуется: французский фармацевтический гигант от него отказался ввиду наличия более совершенных и удобных в использовании GLP1R-агонистов других фармпроизводителей, включая вышеперечисленные.
Болезнь Паркинсона — распространенное, изнуряющее и инвалидизирующее нейродегенеративное заболевание. Тремор в покое, ригидность конечностей, медлительность движений — вот ее наиболее известные признаки, которым сопутствуют осложнения в виде вегетативных симптомов, нарушения сна и когнитивных расстройств. Неуклонное прогрессирование болезни Паркинсона отражается постепенно нарастающей инвалидизацией, справиться с которой не под силу никакому из существующих фармакологических методов лечения [1]. Разработка нейропротекторных методов лечения, способных замедлить, остановить или обратить вспять нейродегенерацию при болезни Паркинсона, уже давно является приоритетной задачей [2].
В 1817 году Джеймс Паркинсон (James Parkinson) в своем «Эссе о дрожательном параличе» оптимистично писал, что, хотя природа болезни ему неизвестна, «существуют достаточные основания надеяться, что в скором времени будет открыт некий лечебный процесс, с помощью которого, по крайней мере, можно будет остановить прогрессирование болезни» [3]. Прошло более двухсот лет, а мы всё еще ждем этого открытия.
Бунтанетап действует сразу на три нейротоксичных белка, ответственных за нейродегенеративные нарушения, — бета-амилоид, альфа-синуклеин и тау-белок.
ЧТО ВЫЯСНИЛОСЬ
Клиническое исследование LixiPark (NCT03439943) фазы II (рандомизированное, двойное слепое, плацебо-контролируемое, многоцентровое) охватило французских пациентов (n=156) в возрасте 40–75 лет с ранней болезнью Паркинсона (стадия < 3 по Хён и Яру), которые придерживались стандартной противопаркинсонической дофаминергической терапии и которым дополнительно на протяжении 12 месяцев ежедневно назначали подкожные инъекции ликсисенатида (lixisenatide) или плацебо.
Первичная конечная точка эффективности лечения была установлена изменением балла в части III унифицированной рейтинговой шкалы оценки болезни Паркинсона Международного общества изучения двигательных расстройств (MDS-UPDRS III). В группе ликсисенатида это изменение составило −0,04 балла, свидетельствуя об улучшении моторных функций, — против +3,04 балла, указывая на ухудшение статуса инвалидизации. Статистически значимая разница составила 3,08 балла (95% ДИ [здесь и далее]: 0,86–5,30; p=0,007) [1].
По прошествии 2-месячного отмывочного периода расхождение сохранилось, причем даже в те моменты, когда пациенты не принимали никаких противопаркинсонических препаратов: усредненный балл получился равным 17,7 пункта (15,7–19,7) — против 20,6 (18,5–22,8) [меньше — лучше].
Применение ликсисенатида сопровождалось тошнотой и рвотой у 46% и 13% испытуемых соответственно.
СУТЬ
GLP1R-агонист ликсисенатид оказал умеренно выраженное благотворное влияние на сдерживание прогрессирования двигательной инвалидизации при болезни Паркинсона, что, возможно, связано с оказываемым им нейропротекторным действием. Предположительно, положительное действие ликсисенатида особенно хорошо себя проявит при возрасте не старше 60 лет и заболевании на относительно ранней стадии. Впрочем, нельзя исключать симптоматического эффекта: доклиническая и клиническая проверка GLP1R-агониста эксенатида (exenatide) при алкогольной и кокаиновой зависимости показала, что он повышает уровень синаптического дофамина [1] [2].
ОДНАКО
Ограниченность клинического испытания лечения болезни Паркинсона GLPR1-агонистом ликсисенатидом не позволяет установить, сохранится ли оказываемый на моторные функции положительный эффект препарата при более длительном его применении или более тяжелой форме заболевания. Неизвестна также величина эффекта большей или меньшей дозы ликсисенатида.
КАК ЭТО РАБОТАЕТ
На данном этапе существуют лишь догадки, что GLP1R-агонисты помогают в лечении болезни Паркинсона, если судить по набору релевантных научных наработок. Так, сахарный диабет 2-го типа является фактором риска развития болезни Паркинсона [1], а его лечение GLP1R-агонистами ассоциировано со снижением этого риска более чем на 50% [2]. На животных моделях болезни Паркинсона GLP1R-агонисты продемонстрировали нейропротекторное действие [3]. В ответ на активацию GLP1R наблюдаются различные физиологические эффекты, в том числе уменьшение воспаления в головном мозге — процесса, который занимает центральной место в патофизиологии болезни Паркинсона [4]. Есть мнение, что активация GLP1R стимулирует нейрогенез и защищает нейроны от опосредованного цитокинами апоптоза, в том числе путем предотвращения микроглиального преобразования астроцитов в нейротоксичный фенотип [3] [5].
Дофаминомиметик тавападон предназначен для улучшения моторных функций при паркинсонизме без обременяющих побочных эффектов.
ЧТО ДАЛЬШЕ
Большинство пациентов с болезнью Паркинсона беспокоит не их нынешнее состояние, а страх прогрессирования моторных нарушений. Если улучшение двигательных функций на 3 балла по шкале MDS-UPDRS III — тот максимум, которого можно добиться от GLP1R-агонистов, то ценность подобного лечения незначительна, особенно с учетом обременительных нежелательных явлений со стороны желудочно-кишечного тракта. Однако если польза такой терапии проявит кумулятивный, накопительный характер, к примеру, добавляя по 3 балла каждый год в течение 5–10 лет и дольше, тогда можно смело говорить о появлении первого в мире лечения, преобразующего и изменяющего течение болезни Паркинсона. Необходимы соответствующие долгосрочные клинические испытания [1].
В БЛИЖАЙШЕМ БУДУЩЕМ
Во второй половине 2024 года ситуация с лечением болезни Паркинсона при помощи GLP1R-агонистов станет более ясной, когда будут готовы результаты клинического исследования Exenatide-PD3 (NCT04232969) фазы III, в котором GLP1R-агонист эксенатид, коммерциализированный «АстраЗенека» (AstraZeneca) как противодиабетический «Бидуреон» (Bydureon), на протяжении 2 лет назначается еженедельными подкожными инъекциями пациентам (n=194) в возрасте 25–80 лет с ранней болезнью Паркинсона (стадия ≤ 2,5 по Хён и Яру), придерживающихся стандартной противопаркинсонической терапии [1].
Итоги Exenatide-PD3 оказались разочаровывающими: не обнаружено статистически значимой разницы между эксенатидом и плацебо в том, что касается сдерживания прогрессирующего ухудшения моторных функций при болезни Паркинсона [2]. Исследователи Университетского колледжа Лондона (University College London, UCL, Лондон, Великобритания) продолжают выяснять, почему эксенатид не справился с поставленной задачей — вопреки ликсисенатиду, который был эффективен.
РАНЕЕ
Предшествовавшие клинические исследования эксенатида, а также экспериментального NLY01, пегилированной версии эксенатида авторства «Ньюрали» (Neuraly), выдали неоднозначные результаты лечения болезни Паркинсона, зависящие от особенностей пациентов [1] [2] [3].
Фармацевтическая отрасль продолжает упорную борьбу с распространенным нейродегенеративным заболеванием.
И ЕЩЁ
Южнокорейская «Пептрон» (Peptron) вынашивала грандиозные планы на экспериментальный PT320 — рецептуру эксенатида с замедленным высвобождением, которая сделана по фирменной технологии SmartDepot на базе биоразлагаемых полимерных микросферических носителей и которая позволяет назначать препарат подкожными инъекциями один раз в две недели [1] [2] [3] [4]. Однако клиническое испытание NCT04269642 фазы II среди пациентов с ранней болезнью Паркинсона, начатое весной 2020 года, так и не завершилось.
ТЕМ ВРЕМЕНЕМ
GLP1R-агонисты продолжают демонстрировать свою пользу за пределами исключительно сахарного диабета 2-го типа и ожирения.
Так, продвигаемый «Ново Нордиск» (Novo Nordisk) семаглутид (semaglutide) доказал, что, во-первых, снижает риск неблагоприятных исходов при сердечно-сосудистом заболевании на фоне ожирения и, во-вторых, успешно справляется с лечением сердечной недостаточности с сохраненной фракцией выброса (HFpEF) среди пациентов с ожирением. Впрочем, это было предсказуемо, учитывая, насколько лишний вес токсичен для сердечно-сосудистой системы.
Эффективное снижение веса при помощи семаглутида сопровождается снижением риска сердечно-сосудистой смерти, инфаркта миокарда и инсульта.
Семаглутид также смог сдержать прогрессирование хронической болезни почек и снизить риск сердечно-сосудистой и почечной смерти у пациентов с сахарным диабетом 2-го типа [1].
Летом 2024 года «Илай Лилли» (Eli Lilly) расскажет, насколько «Зепбаунд» (Zepbound, тирзепатид) терапевтически востребован в случае HFpEF с сопутствующим ожирением: этот вопрос раскрывается в клиническом исследовании SUMMIT (NCT04847557) фазы III.
К осени 2025 года станет известно, пригоден ли семаглутид в лечении болезни Альцгеймера: способен ли «Ребелсас» (Rybelsus), принимаемый ежедневно перорально на протяжении 2 лет в рамках клинического испытания EVOKE (NCT04777396) фазы III, замедлить прогрессирование деменции у пациентов с ранней болезнью Альцгеймера.
Семаглутид против ожирения попутно ослабит бремя сердечной недостаточности с сохраненной фракцией выброса.
Весной 2026 года ожидаются результаты клинического исследования GALLOP (NCT05920889) фазы II, которое проверяет гипотезу, что добавление семаглутида к стандартной механической процедуре эндоваскулярной тромбэктомии (EVT) при остром ишемическом инсульте, вызванном окклюзией крупных сосудов, предотвращает неблагоприятные исходы, обусловленные перипроцедурным злокачественным отеком мозга (MBE) и симптоматическим внутричерепным кровоизлиянием (sICH).
Тема неалкогольного стеатогепатита (НАСГ), интересующая буквально каждого игрока «Большой фармы» ввиду огромных бизнес-перспектив, но по факту остающаяся без действительно сильных лекарств, прорабатывается в клиническом исследовании ESSENCE (NCT04822181) фазы III, в котором изучается длительное (максимум 5 лет) еженедельное применение инъекционного семаглутида при НАСГ без цирроза печени и с ее фиброзом на стадии F2–F3.
Продолжается тестирование семаглутида среди людей с коморбидным ожирением и алкогольной зависимостью: по силам ли «Вегови» (Wegovy), который в ходе клинического исследования SEMALCO (NCT05895643) фазы II назначается еженедельными подкожными инъекциями, снизить потребление алкоголя или даже избавить от алкоголизма.
«Ново Нордиск» (Novo Nordisk) осуществила два больших шага к удовлетворению высочайшего общемирового спроса на свои лекарства-бестселлеры на основе семаглутида (semaglutide), агониста рецептора глюкагоноподобного пептида-1 (GLP1RA), — препараты «Оземпик» (Ozempic) против сахарного диабета 2-го типа (СД2) и «Вегови» (Wegovy) для похудения.
Датский фармпроизводитель, во-первых, выделил 8,5 млрд крон ($1,2 млрд) на строительство нового завода в родной Дании [1] и, во-вторых, готов закрыть сделку по покупке за $11,7 млрд трех производственных площадок за рубежом [2].
Новый производственный комплекс и складские помещения, которые к 2027 году будут возведены на окраине Оденсе, третьего по величине города Дании, займут 40 тыс. кв. м. Завод, расположенный в двух часах езды от Копенгагена, проектируется по «модульному и гибкому принцу» и рассчитан на выпуск не только GLP1RA-препаратов, но и прочих инъекционных лекарственных средств.
В середине декабря 2014 года Федеральная торговая комиссия США (FTC) уведомила, что не собирается оспаривать приобретение контрактного производителя «Каталент» (Catalent), о котором «Ново Нордиск» объявила в начале февраля [3]. Ранее, в начале декабря, Европейская комиссия одобрила покупку [4].
Согласно условиям трехсторонней сделки, после того как «Ново Холдингс» (Novo Holdings), крупнейший (контролирует 77% голосов) акционер «Ново Нордиск», закроет покупку «Каталент» за $16,5 млрд, затем три завода (два в Европе и один в США) последней будут проданы «Ново Нордиск» за $11,7 млрд [5].
Поглощение «Каталент» встретило тщетное сопротивление в США: группы защиты потребителей, профсоюзные организации и сенатор Элизабет Уоррен (Elizabeth Warren) выказали опасения, что это приведет к затруднениям у других фармпроизводителей с разработкой и выводом на рынок собственных препаратов для похудения. Как утверждается, такие фармкомпании, как «Амджен» (Amgen), «Пфайзер» (Pfizer), «Рош» (Roche), «АстраЗенека» (AstraZeneca), «Вайкинг терапьютикс» (Viking Therapeutics), «Стракча терапьютикс» (Structure Therapeutics), столкнутся с затруднениями с доступом к фабричным площадкам, необходимым для выпуска инъекционных лекарственных форм, поскольку все заводы оккупирует «Ново Нордиск» [6] [7].
Помимо вышеуказанных приобретений «Ново Нордиск» за минувших полтора года сообщила об инвестициях в размере $14,92 млрд, которые аналогично будут пущены на расширение производственных мощностей: в США, Дании и Франции [8] [9].
Ажиотажный спрос на GLP1RA-препараты для быстрого и относительно беспроблемного снижения лишнего веса вынуждает «Ново Нордиск» вкладывать всё больше средств, чтобы удовлетворить растущие потребности пациентов, тем паче лекарственные средства этого класса продолжают демонстрировать терапевтические преимущества далеко за пределами лечения СД2 и ожирения: к примеру, в терапии метаболически ассоциированного стеатогепатита (МАСГ), алкоголизма, болезни Паркинсона. Датский фармацевтический гигант не жалеет ресурсов на осуществление клинических проверок как семаглутида, так и экспериментального его сочетания с кагрилинтидом (cagrilintide), аналогом амилина длительного действия, — массив испытаний охватывает свыше 45 тыс. человек [10].
В январе 2024 года Алессандро Мазелли (Alessandro Maselli), исполнительный директор «Каталент», сообщил акционерам, что доход предприятия от контрактного выпуска GLP1RA-препаратов превысит $500 млн [11].
Если весной 2023 года отраслевые эксперты прогнозировали, что мировой рынок современных лекарств для похудения доберется до $100 млрд в начале 2030-х гг., то уже через год, весной 2024-го, предсказания оптимистично выросли до $150 млрд [12] [13].
«Ново Нордиск», которая на дату середины декабря 2024 года, оставалась второй крупнейшей в мире фармацевтической компанией с рыночной капитализацией $491 млрд, уступая лишь «Илай Лилли» (Eli Lilly) [$710 млрд], чрезвычайно сильным образом влияет на всю экономику родной Дании, меняя ее традиционные социальные устои и определяя переток рабочей силы [14].
«Илай Лилли», напрямую конкурирующая с «Ново Нордиск» благодаря своим лекарственным препаратам на основе тирзепатида (tirzepatide), одновременно GLP1RA и агониста рецептора глюкозозависимого инсулинотропного полипептида (GIPRA), времени не теряет. На волне такого же сумасшедшего спроса на «Мунджаро» (Mounjaro) против СД2 и «Зепбаунд» (Zepbound) против ожирения американский фармпроизводитель пытается нарастить производственные мощности. В конце мая 2024 года было объявлено об увеличении инвестиций, которые пойдут на расширение фабричных площадей: с $3,7 млрд до $9 млрд [15].
Касательно динамики продаж лекарств на базе семаглутида и тирзепатида, картина такова, что в 2022 и 2023 гг. совокупная реализация «Оземпика» и «Вегови» составила $9,31 млрд и $18,44 млрд, тогда как общая торговля «Мунджаро» и «Зепбаундом» вышла к $483 млн и $5,34 млрд. За первых девять месяцев 2024 года эти препараты «Ново Нордиск» сгенерировали $18,15 млрд, а лекарства «Илай Лилли» — $11,03 млрд.
Получается, что разрыв между объемами продаж сокращается, стремительно и неумолимо: ×19,3 ➞ ×3,5 ➞ ×1,6 — во столько раз заработок датской фармкомпании на семаглутиде превышал заработок американской на тирзепатиде в указанные одинаковые периоды времени. Если тенденция продолжится, продажи препаратов на основе тирзепатида в обозримом будущем опередят таковые для лекарств на базе семаглутида.
«Анлоксит» (Unloxcyt, косибелимаб) — новый лекарственный препарат, предназначенный для лечения плоскоклеточного рака кожи.
ОСНОВНЫЕ ФАКТЫ
«Анлоксит» ориентирован на лечение взрослых пациентов с плоскоклеточной карциномой кожи: либо метастатической, либо местнораспространенной и неподходящей для радикальных хирургии или облучения.
Косибелимаб (cosibelimab), разработанный «Чекпойнт терапьютикс» (Checkpoint Therapeutics), представляет собой блокатор PD-L1.
«Анлоксит» одобрен Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) в середине ноября 2024 года [1].
Косибелимаб — прямой конкурент PD-1-блокаторов «Либтайо» (Libtayo, цемиплимаб) и «Китруда» (Keytruda, #пембролизумаб) авторства «Ридженерон фармасьютикалс» (Regeneron Pharmaceuticals) / «Санофи» (Sanofi) и «Мерк и Ко» (Merck & Co.), которые уже применяются в лечении плоскоклеточного рака кожи.
Эффективность косибелимаба не уступает таковой у цемиплимаба (cemiplimab) и пембролизумаба (pembrolizumab), зато профиль безопасности всё же лучше.
СУТЬ ВОПРОСА
Плоскоклеточная карцинома кожи (ПККК), или плоскоклеточный рак кожи (ПКРК) — вторая по распространенности форма рака кожи: на него приходится приблизительно 20% эпителиальных раков кожи [1] [2] [3].
Заболеваемость ПКРК быстро растет во всём мире в связи со старением населения планеты и усилением воздействия факторов риска, таких как ультрафиолетовое излучение [1] [4] [5]. Так, согласно исследованию образца 2020 года, в период 2017–2027 гг. уровень заболеваемости плоскоклеточным раком кожи в Европе вырастет на 23% и 29% — соответственно среди мужчин и женщин [4]. Аналогичным образом растет число случаев ПКРК в США, Великобритании, Австралии [1] [6] [7]. Всё это отразится соответствующим увеличением смертности и усилением бремени на системы общественного здравоохранения [3].
Тем не менее плоскоклеточный рак кожи остается недостаточно признанным заболеванием, поскольку исключен из большинства национальных онкологических регистров [8].
У пациентов с ПКРК обычно отмечается благоприятный прогноз с высокой вероятностью долгосрочного выживания после хирургического иссечения, которое остается стандартным методом лечения локализованного плоскоклеточного рака кожи [3].
Приблизительно 17% иммунокомпетентных пациентов сталкиваются с локорегионарным рецидивом в течение пяти лет после хирургического вмешательства и послеоперационной лучевой терапии [9]. В случаях рецидива, агрессивного прогрессирования или развития местнораспространенной или метастатической плоскоклеточной карциномы кожи, ассоциированных со значительной заболеваемостью и смертностью, вариантов лечения не так много [1] [3] [10], притом что метастазирование сокращает до 10–20% шансы выжить на протяжении десяти лет [11] [12].
Парадигма лечения плоскоклеточного рака кожи кардинальным образом поменялась с появлением иммунотерапии.
В конце сентября 2018 года на сцену вышел Либтайо» (Libtayo, цемиплимаб) — первый специфический препарат для первоочередной терапии метастатической или местнораспространенной плоскоклеточной карциномы кожи. В конце января 2020-го лечение рецидивирующего или метастатического плоскоклеточного рака кожи подключил «Китруда» (Keytruda, пембролизумаб), в начале июля 2021 года добавивший терапию местнораспространенного заболевания.
Эти блокаторы PD-1, разработанные соответственно «Ридженерон фармасьютикалс» (Regeneron Pharmaceuticals) / «Санофи» и «Мерк и Ко» (Merck & Co.), прекрасно справляются с поставленной задачей благодаря тому, что плоскоклеточная карцинома кожи характеризуется исключительно высоким опухолевым мутационным бременем ввиду ультрафиолетового излучения как основного фактора риска заболевания [13].
PD-L1, который высоко экспрессирован на опухолевых клетках плоскоклеточной карциномы кожи, является основным лигандом для PD-1 [14]. Активация PD-1 на Т-клетках, индуцированная PD-L1 на раковых клетках, приводит к подавлению выработки цитокинов и цитолитической активности инфильтрирующих опухоль Т-клеток PD-1+ — опухолевые клетки избегают уничтожения иммунной системой [10] [14] [15] [16]. Кроме того, повышенная опухолевая экспрессия PD-L1 ассоциирована с риском метастазирования [17]. Терапевтическое ингибирование взаимодействия между PD-1 и PD-L1 активирует иммунный ответ и приводит к противоопухолевой активности [10].
И хотя моноклональные антитела, блокирующие PD-1, высокоэффективны в лечении плоскоклеточного рака кожи, их применение несет за собой риск серьезных нежелательных явлений (НЯ), что указывает на необходимость поиска новых препаратов с улучшенным профилем безопасности [18].
Отмечены различия между блокаторами PD-1 и блокаторами PD-L1 в частоте возникновения связанных с лечением НЯ (TRAE), проявляющихся в тяжелой или жизнеугрожающей форме, включая иммуноопосредованные НЯ (irAE), причем частота их возникновения ниже у блокаторов PD-L1 [18] [19] [20].
Существует гипотеза, что при применении антител против PD-L1 частота TRAE и irAE может быть ниже, поскольку взаимодействие между PD-1 и PD-L2 остается неизменным: при более слабом подавлении отрицательного ингибирующего сигнала (благодаря сохранению сигнализации PD-L2) блокаторы PD-L1 могут вызывать меньший аутоиммунитет по сравнению с антителами против PD-1 [18] [20]. Кроме того, PD-L2 также связывается с молекулой отталкивающего наведения b (RGMb, DRAGON), которая регулирует респираторный иммунитет; этот факт, возможно, объясняет, почему частота некоторых irAE, таких как пневмонит, выше при использовании блокаторов PD-1, чем блокаторов PD-L1 [21] [22].
«Анлоксит» (Unloxcyt, косибелимаб) стал первым блокатором PD-L1, одобренным в лечении плоскоклеточного рака кожи.
КЛИНИЧЕСКИЕ ПОДРОБНОСТИ
Клиническое исследование NCT03212404 фазы I (нерандомизированное, открытое, многокогортное, многоцентровое, международное) охватило взрослых пациентов (n=109) с плоскоклеточной карциномой кожи, либо метастатической, либо местнораспространенной и неподходящей для радикальных хирургии или облучения.
Участникам назначали внутривенные инфузии косибелимаба каждые две недели — до момента подтвержденного полного ответа, прогрессирования заболевания или неприемлемой токсичности.
По прошествии медианных 15,4 месяца (95% ДИ [здесь и далее]: 12–21) наблюдений результаты получились следующими [1]:
частота полного ответа (ORR): для метастатического заболевания — 47% (36–59), включая 8% полных ответов (CR) и 40% частичных ответов (PR), для местнораспространенного — 48% (30–67), в том числе CR 10% и PR 39%;
медиана длительности ответа (DOR): не достигнута (1,4+ — 34,1+) и 17,7 месяца (3,7+ — 17,7+);
пропорция пациентов с DOR на протяжении 6 месяцев: 84% и 87%;
пропорция пациентов с DOR на протяжении 12 месяцев: 54% и 20%.
После наблюдений на протяжении медианных 33 и 24 месяцев за пациентами с метастатическим или местнораспространенным плоскоклеточным раком кожи результаты лечения улучшились: частоты ORR выросли до соответствующих 50% (39–62%) и 55% (36–73), включая CR 13% и 26%. Медиана DOR всё еще достигнута не была.
ЭКСПЕРТНЫЕ КОММЕНТАРИИ
«Анлоксит» (Unloxcyt, косибелимаб) авторства «Чекпойнт терапьютикс» (Checkpoint Therapeutics), будучи блокатором PD-L1, вошел в прямое противостояние с двумя блокаторами PD-1: «Либтайо» (Libtayo, цемиплимаб) и «Китрудой» (Keytruda, пембролизумаб), продвигаемых соответственно «Ридженерон фармасьютикалс» (Regeneron Pharmaceuticals) / «Санофи» (Sanofi) и «Мерк и Ко» (Merck & Co.) и одобренных при таких же показаниях лечения метастатического или местнораспространенного плоскоклеточного рака кожи.
Если сравнивать терапевтическую эффективность косибелимаба (cosibelimab) с цемиплимабом (cemiplimab) и пембролизумабом (pembrolizumab), хотя это методологически неверно, картина рисуется следующей.
Назначение цемиплимаба (n=193) при метастатическом заболевании обеспечило ORR 46% (CR 20% и PR 27%), медиану DOR 41 месяц и 12-месячную DOR у 88% пациентов, при местнораспространенном — ORR 45% (CR 13% и PR 32%), медиану DOR 42 месяца и 12-месячную DOR у 69% больных [1] [2] [3].
Применение пембролизумаба при рецидивирующем или метастатическом заболевании (n=150) привело к ORR 36% (CR 12% и PR 23%), без достижения медианы DOR, 12-месячной DOR у 68% человек, а при местнораспространенном заболевании (n=54) — ORR 52% (CR 22% и PR 30%), медиане DOR 47,2 месяца, 12-месячной DOR у 75% испытуемых [4].
Таким образом, блокада PD-L1 или PD-1 одинаково эффективно справляется с лечением плоскоклеточной карциномы кожи.
Что касается связанных с лечением нежелательных явлений (TRAE), профиль безопасности явно складывается в пользу косибелимаба. Так, использование препарата привело в основном к легко-умеренным TRAE: только у 10% пациентов были зарегистрированы TRAE в тяжелой форме, случаев жизнеугрожащих или летальных TRAE не зафиксировано. С иммуноопосредованными нежелательными явлениями (irAE) столкнулись 23% испытуемых, причем в тяжелой форме они были отмечены лишь у 3% человек. Всё это выглядит лучше, чем ситуация с TRAE и irAE в аналогичных клинических испытаниях блокаторов PD-1 — цемиплимаба и пембролизумаба [1] [2] [3] [4].
Экспериментальный препарат бепировирсен (bepirovirsen), разрабатываемый «ГлаксоСмитКляйн» (GlaxoSmithKline), располагает необходимым потенциалом, для того чтобы обеспечить функциональное излечение хронического вирусного гепатита B.
ОСНОВНЫЕ ФАКТЫ
Продемонстрировано, что еженедельные подкожные инъекции бепировирсена привели к полному клиренсу ДНК и поверхностного антигена вируса гепатита В (HBsAg) у почти трети пациентов после 6 месяцев лечения.
Впрочем, по прошествии 6 месяцев наблюдений после завершения терапии статус функционального излечения оказался справедливым для существенно меньшей пропорции больных. Однако терапию всё равно можно назвать успешной, если сравнивать с существующими лекарственными средствами.
В настоящее время хронический вирусный гепатит B, которым заражены 254 млн человек во всём мире, неизлечим: приходится придерживаться пожизненной терапии, которая далеко не всегда оказывается успешной [1].
Напротив, хронический вирусный гепатит C считается полностью излечимым заболеванием. С мая 2011 года стали планомерно появляться всё более эффективные противовирусные препараты прямого действия (ПППД), за несколько месяцев исцеляющие эту инфекционную болезнь.
Цена лекарств постоянно растет, несмотря на отсутствие значимых улучшений. Разбираемся, почему фармацевтическая индустрия превратилась в машину для выкачивания денег.
КАК ЭТО РАБОТАЕТ
Бепировирсен (bepirovirsen, GSK3228836, IONIS-HBVRx) представляет собой антисмысловой олигонуклеотид (ASO) — одноцепочечную ДНК, которая комплементарна 20 консервативным нуклеотидным последовательностям всех матричных РНК (мРНК) вируса гепатита B (HBV), включая его прегеномную РНК (пгРНК).
Связывание бепировирсена с мРНК и пгРНК HBV приводит к образованию гибридного комплекса, который рекрутирует эндогенную рибонуклеазу H (РНКаза H). Этот фермент расщепляет мРНК и пгРНК HBV, тем самым препятствуя трансляции белков вируса. В результате уменьшается количество РНК, ДНК и белков HBV, в том числе поверхностного антигена вируса гепатита В (HBsAg). Вирусная нагрузка снижается, сдерживаются процессы инфицирования и репликации HBV, появляется шанс на функциональное излечение хронического вирусного гепатита B [1] [2].
Бепировирсен также проявляет агонистическую активность в отношении толл-подобного рецептора 8 (TLR8), тем самым работая как иммуностимулятор, индуцирующий активность врожденного иммунитета и выработку цитокинов [3] [4] [5] [6] [7].
Поскольку бепировирсен не конъюгирован с аминосахаром N-ацетилгалактозамином (GalNAc), нужным для более таргетной доставки в печень, он в большей степени распределяется в непаренхимных клетках печени, нежели в гепатоцитах, и потому распознается резидентными иммунными клетками печени, что приводит к активации сигнализации врожденной иммунной системы [8].
Бепировирсен разработан «Айонис фармасьютикалс» (Ionis Pharmaceuticals), которая в конце августа 2019 года лицензировала его «ГлаксоСмитКляйн» (GlaxoSmithKline). Взамен обещано до 262 млн долларов и роялти от реализации готового лекарственного препарата [9].
Состоятельность терапевтической парадигмы антисмысловых олигонуклеотидов подтверждена немалым числом уже одобренных лекарственных препаратов, направленных на сдерживание экспрессии специфических белков, патогенных в случае какого-либо заболевания. Так, например, «Эксондис 51» (Exondys 51, этеплирсен), «Виондис 53» (Vyondys 53, голодирсен), «Амондис 45» (Amondys 45, касимерсен) и «Вилтепсо» (Viltepso, вилтоларсен) применяются в лечении мышечной дистрофии Дюшенна, «Спинраза» (Spinraza, нусинерсен) используется в лечении спинальной мышечной атрофии, «Тегседи» (Tegsedi, инотерсен) назначается для лечения полинейропатии при наследственном транстиретиновом амилоидозе.
КЛИНИЧЕСКИЕ ПОДРОБНОСТИ
В целях предварительного выяснения эффективности и безопасности лечения хронического вирусного гепатита B при помощи бепировирсена «ГлаксоСмитКляйн» положилась на клиническую программу из трех испытаний фазы II:
B-Clear (NCT04449029): 24- или 12-недельное назначение бепировирсена (разными схемами), причем либо на фоне терапии нуклеозидными/нуклеотидными аналогами (NA), либо без нее.
B-Together (NCT04676724): 24- или 12-недельное применение бепировирсена на фоне NA-терапии, за которым следует 24-недельный курс пегилированного интерферона альфа-2a.
Первичная конечная точка эффективности лечения хронического вирусного гепатита B в первых двух клинических испытаниях была установлена пропорцией пациентов, продемонстрировавших устойчивую вирусную супрессию (подавление вирусной нагрузки; SVR), под которой понимают снижение уровней поверхностного антигена вируса гепатита В (HBsAg) и ДНК HBV ниже определяемых высокоточным методом ПЦР порогов (соответственно 0,05 МЕ/мл и 20 МЕ/мл), сохраняющееся на протяжении 24 недель после завершения лечения и при условии отсутствия в этот период дополнительной терапии какими-либо противовирусными препаратами (если таковые ранее не принимались).
B-Sure (NCT04954859): среди ранее прошедших лечение бепировирсеном и ответивших на него пациентов из других клинических испытаний. В ходе 33-месячных наблюдений выясняется, как долго сохраняется SVR-статус.
B-CLEAR
Клиническое исследование B-Clear (NCT04449029) фазы IIb (рандомизированное, двойное слепое, плацебо-контролируемое, многоцентровое, международное) пригласило взрослых пациентов (n=457) с хроническим вирусным гепатитом B, проходящих либо нет фоновое лечение нуклеозидными/нуклеотидными аналогами (NA).
На протяжении 24 недель участникам еженедельно подкожными инъекциями назначали бепировирсен (разными схемами):
группа 1: бепировирсен 300 мг — 24 недели;
группа 2: бепировирсен 300 мг — 12 недель, затем бепировирсен 150 мг — 12 недель;
группа 3: бепировирсен 300 мг — 12 недель, затем плацебо — 12 недель;
группа 4: плацебо — 12 недель, затем бепировирсен 300 мг — 12 недель.
Группы 1, 2 и 3 также получили нагрузочные дозы бепировирсена: по 300 мг на 4-й и 11-й дни.
Согласно промежуточному анализу собранных данных, после 24-недельной терапии хронического вирусного гепатита B наилучшую результативность показала группа 1. Так, одновременное отсутствие HBsAg и ДНК HBV зафиксировано для 28% и 29% испытуемых, соответственно придерживавшихся фоновой терапии NA и не принимавших такие препараты. При этом у 68% и 65% участников уровень HBsAg упал ниже 100 МЕ/мл [1].
Итоговые результаты клинического исследования получились следующими [2].
Среди тех пациентов, которые придерживались фоновой терапии NA, к первичной конечной точке эффективности лечения вышли 9%, 9%, 3% и 0% участников в группах 1, 2, 3 и 4. Среди тех, кто не получал NA, первичная конечная точка достигнута среди 10%, 6%, 1% и 0% испытуемых.
Если допустить «всплески» активности вируса гепатита B (однократное повышение HBsAg или ДНК HBV до уровней, превышающих или равных пороговым), то есть несколько ослабить критерии выхода к первичной конечной точке, ее достигли 10%, 9%, 4% и 2% пациентов на фоновой терапии NA и 14%, 6%, 1% и 4% пациентов без фоновой терапии NA.
Успешный ответ на назначение бепировирсена напрямую зависел от исходного уровня HBsAg до начала лечения: он чаще фиксировался у испытуемых с низким уровнем HBsAg (≤ 3 log10 МЕ/мл) и реже с высоким (> 3 log10 МЕ/мл). К примеру, в группе 1 первичная конечная точка была засвидетельствована у 16% и 25% пациентов с низким изначальным уровнем HBsAg и у 6% и 7% пациентов с высоким — соответственно среди получавших и не получавших фоновую терапию NA.
Бепировирсен характеризовался приемлемой переносимостью. Наиболее распространенным нежелательным явлением были реакции по месту введения препарата.
B-TOGETHER
Клиническое исследование B-Together (NCT04676724) фазы IIb (рандомизированное, открытое, многоцентровое, международное) пригласило взрослых пациентов (n=108) с хроническим вирусным гепатитом B.
Испытуемым, продолжающим следовать стабильной терапии нуклеозидными/нуклеотидными аналогами (NA), назначали бепировирсен (еженедельными подкожными 300-мг инъекциями) на протяжении 24 недель (группа 1) или 12 недель (группа 2); плюс нагрузочные дозы бепировирсена (по 300 мг на 4-й и 11-й дни). Далее участники проходили 24-недельный курс пегилированного интерферона альфа-2a (в еженедельной подкожной дозе 180 мкг).
Экспериментальное лечение обеспечило выход к первичной конечной точке для 9% и 15% пациентов в группах 1 и 2. При этом исходный уровень HBsAg коррелировал с пропорцией ответивших на лечение больных. Так, при низком изначальном уровне HBsAg (≤ 1000 МЕ/мл) первичная конечная точка была зафиксирована для 24% и 41% испытуемых, тогда как при высоком его уровне (≤ 3000 МЕ/мл) — для 14% и 26% [1].
Большинство пациентов (58% в каждой группе), показавших ответ на лечение по завершении применения бепировирсена, не столкнулись с рецидивом инфекции в ходе назначения интерфероновой терапии. Другими словами, добавление интерферона снизило риск рецидива хронического вирусного гепатита B.
Однако только 2 человека (в группе 2) с частичным ответом после бепировирсена вышли к полному ответу в процессе добавления интерферона. То есть подключение последнего к бепировирсену не привело к значимому улучшению исходов, обусловленных снижением уровня HBsAg.
Был отмечен временный рост уровня АЛТ выше утроенной верхней границы нормы. В ходе интерфероновой терапии не было отмечено ассоциации между ростом HBsAg и АЛТ.
Серьезные нежелательные явления (НЯ), связанные с лечением, были зарегистрированы только у 1 пациента (2%). Некоторые НЯ вынудили выйти из исследования 4% испытуемых (n=4): аллергический дерматит, реакции по месту введения препарата, депрессия.
B-SURE
Продолжающееся клиническое испытание B-Sure (NCT04954859) фазы II (нерандомизированное, открытое, многоцентровое, международное) поставило своей целью выяснить, как долго сохраняется SVR-статус среди ранее прошедших лечение бепировирсеном и ответивших на него пациентов с хроническим вирусным гепатитом B из других клинических испытаний этого экспериментального препарата. Наблюдательное исследование продолжается сроком максимум 33 месяца.
Положительный SVR-статус разнесен по критерию ответа на бепировирсен: полный ответ (CR) и частичный ответ (PR). Первый, отражающий функциональное излечение хронического вирусного гепатита B, предполагает снижение уровней поверхностного антигена вируса гепатита В (HBsAg) и ДНК вируса гепатита B (HBV) ниже порогов, определяемых высокоточным ПЦР-методом: соответственно 0,05 МЕ/мл и 20 МЕ/мл. Второй — уровень HBsAg < 100 МЕ/мл и уровень ДНК HBV ниже вышеуказанного порогового.
Если говорить о пациентах из B-Clear (NCT04449029), то анализ исходов B-Sure осуществляется согласно разбивке по факту терапии нуклеозидными/нуклеотидными аналогами (NA): одна группа пациентов из B-Clear получала фоновое NA-лечение, но затем, по прошествии 3 месяцев после начала участия в B-Sure, должна была прекратить, тогда как вторая группа вообще не проходила фоновую NA-терапию.
В группе фонового NA-лечения оказались 11 человек с полным ответом, из которых 9 пациентов впоследствии, согласно протоколу исследования, прекратили прием NA-препаратов. По прошествии 6 месяцев после остановки NA-терапии полный ответ сохранился у 78% (n=7/9) участников. По прошествии еще 6 месяцев (всего 12), когда 1 больной был исключен из анализа ввиду недоступности данных наблюдений за ним, полный ответ сохранился у всех оставшихся 6 человек, то есть составил 67% (n=6/9).
Ситуация с пациентами, показавшими частичный ответ к моменту включения в B-Sure, следующая. Из 29 пациентов прием NA-препаратов должным образом прекратили 23 человека. По истечении 6 месяцев частичный ответ сохранился у 22% испытуемых (n=5/23), притом что 13% (n=3/23) продемонстрировали отложенный полный ответ. После 12 месяцев частичный ответ сохранился у 13% (n=3/23), а отложенный полный ответ — у 13% (n=3/23).
В группу, не проходившую фоновое NA-лечение, попали 16 пациентов: 11 человек с полным ответом и 5 с частичным. По прошествии 15 месяцев полный ответ сохранился у 36% (n=4/11), частичный — у 20% (n=1/5).
Таким образом, лечение хронического вирусного гепатита B при помощи бепировирсена способно привести к функциональному излечению этой инфекции, что подтверждается результатами долгосрочных наблюдений за пациентами, прекратившими всякое лечение заболевания.
ЧТО ДАЛЬШЕ
На волне обнадеживающих результатов, продемонстрированных бепировирсеном в задаче излечения хронического вирусного гепатита B, «ГлаксоСмитКляйн» запустила два идентичных опорных клинических испытания, B-Well 1 (NCT05630807) и B-Well 2 (NCT05630820), фазы III (рандомизированных, двойных слепых, плацебо-контролируемых, многоцентровых, международных), которые, если завершатся успешно, лягут в основу регистрационного досье.
Среди основных требований к взрослым участникам (n=900 и n=900): стабильная терапия нуклеозидными/нуклеотидными аналогами (NA) на протяжении не менее чем 6 месяцев; уровень HBsAg в пределах 100–3000 МЕ/мл; уровень ДНК HBV < 90 МЕ/мл; уровень АЛТ не выше удвоенной верхней границы нормы.
Испытуемые, продолжающие следовать фоновой NA-терапии, вначале проходят 24-недельный курс терапии бепировирсеном или плацебо, а затем на протяжении 24 или 48 недель получают только NA-препараты.
Первичная конечная точка эффективности лечения установлена функциональным излечением хронического вирусного гепатита B, факт которого подтверждается устойчивой вирусной супрессией (SVR) на протяжении хотя бы 24-недельного периода, оставляемого без какого-либо лечения. Исследования завершатся ближе к концу 2025 года.
В клиническом исследовании B-United (NCT06537414) фазы IIb пациентам (n=280) с хроническим вирусным гепатитом B, придерживающимся стабильной NA-терапии и находящимся в SVR-статусе, вначале назначают комбинацию из даплусирана (daplusiran) и томлигисирана (tomligisiran) — по 50 и 200 мг каждые 4 недели на протяжении 24 недель, а затем проводят 24-недельный курс лечения бепировирсеном. Результаты испытания, которое должно выяснить частоту функционального излечения инфекции, будут готовы к концу 2027 года.
Комбинация из даплусирана и томлигисирана (JNJ-3989, JNJ-73763989, GSK5637608, ARO-HBV) — фиксированная доза малых интерферирующих РНК (миРНК), таргетированных на гепатоциты и индуцирующих процесс эндогенной интерференции для расщепления транскриптов РНК HBV, экспрессируемых как из ковалентно замкнутой кольцевой ДНК (кзкДНК) HBV, так и из ДНК HBV, интегрированной в геном хозяина. Это приводит к снижению уровня всех белков HBV (HBsAg, HBeAg) и его прегеномной РНК (пгРНК) [1].
Самостоятельно сочетание даплусирана и томлигисирана снижает уровень HBsAg, причем независимо от его исходной концентрации, но обеспечить функциональное излечение хронического вирусного гепатита B не в силах. Антисмысловой олигонуклеотид бепировирсен продемонстрировал свою максимальную эффективность в отношении устойчивой потери HBsAg среди пациентов с изначально относительно низким уровнем HBsAg (≤ 3000 МЕ/мл). Отсюда и родилась гипотеза, что, если снизить уровень последнего перед назначением бепировирсена, можно увеличить пропорцию пациентов, которые выйдут к статусу функционального излечения [2].
В клиническом испытании B-Focus (NCT06497504) фазы II изучается лечение пациентов (n=150) с коинфекцией хронического вирусного гепатита B и вируса иммунодефицита человека 1 (ВИЧ-1). Последний должен находиться в статусе вирусной супрессии благодаря антиретровирусной терапии (АРТ). Результаты будут собраны к середине 2027 года.
ЧТО ЕЩЕ
«ГлаксоСмитКляйн» осуществляет клиническое исследование NCT05276297 фазы II, в котором пациентам (n=184) после 12- или 24-недельной терапии хронического вирусного гепатита B бепировирсеном следует назначение экспериментальной таргетной иммунотерапии GSK3528869A.
Первичная конечная точка эффективности лечения установлена устойчивой вирусной супрессией (SVR) по прошествии 24 недель после терапии. Испытание должно завершиться к зиме 2026 года.
GSK3528869A представляет собой иммунотерапевтическую вакцину из трех компонентов:
ChAd155-hIi-HBV: лишенный возможности реплицироваться аденовирус шимпанзе группы C серотипа 155, кодирующий последовательности двух белковых антигенов HBV: усеченного ядерного антигена вируса гепатита В (HBcAg) и полноразмерного малого поверхностного антигена вируса гепатита В (S-HBsAg);
MVA-HBV: кодирующий два вышеуказанных белковых антигена HBV модифицированный осповакцинный вирус Ankara (Modified vaccinia Ankara, MVA), представляющий собой высокоаттенуированный штамм вируса осповакцины (Vaccinia virus);
HBc-HBs/AS01B-4: вышеуказанные белковые антигены HBV, подкрепленные адъювантом AS01B-4, который представляет собой липосомальное сочетание 3-О-дезацилированного 4′-монофосфорил-липида A (MPL) сальмонеллы (Salmonella minnesota) и молекулы сапонина (QS-21) из растительного экстракта квиллайи мыльной (Quillaja saponaria).
Первый компонент GSK3528869A вводится по завершении курса бепировирсеном: в 1-й день, второй — в 57-й, третий — в 113-й и 169-й.
Концептуальная идея применения терапевтических вакцин вроде GSK3528869A состоит в том, что недостаточность индукции HBV-специфического B- и T-клеточного иммунитета ответственна за отсутствие полного клиренса вируса гепатита B [1] [2] [3] [4]. Вакцина должна запускать формирование сильного вирусоспецифического иммунитета против антигенов HBV, контролирующего инфекцию путем индукции нейтрализующих антител и элиминации инфицированных гепатоцитов при участии эффекторных T-клеток [4] [5].
В начале декабря 2024 года «ГлаксоСмитКляйн» остановила клиническую проверку NCT03866187 фазы I/II иммунотерапевтической вакцины GSK3528869A, назначаемой пациентам (n=135) с хроническим вирусным гепатитом B, находящимся в SVR-статусе благодаря терапии нуклеозидными/нуклеотидными аналогами (NA). Заявлено об отсутствии должной эффективности по прошествии 24 недель после лечения [6].
Клиническое исследование NCT05330455 фазы I/II, которое должно завершиться к концу 2027 года, тестирует среди пациентов (n=132) с хроническим вирусным гепатитом B сочетание из бепировирсена и GSK3965193, низкомолекулярного ингибитора неканонической атипичной поли(А)-полимеразы 5 и 7 (PAPD5 и PAPD7). Эти ферменты нужны для стабилизации РНК HBV посредством вирусного посттранскрипционного регуляторного элемента (PRE) [7] [8] [9]. Ингибирование PAPD5 и PAPD7 приводит к подавлению вирусной репликации и синтеза вирусных белков, включая HBsAg [10] [11] [12]. В доклинических исследованиях на мышиной модели HBV продемонстрирована оправданность комбинации бепировирсена и GSK3965193 с позиции усиления снижения уровня HBsAg [13].
ЭКСПЕРТНЫЕ КОММЕНТАРИИ
Все существующие стратегии лечения хронического вирусного гепатита B преследуют цель долгосрочной супрессии (подавления) уровня ДНК вируса гепатита B (HBV). При этом весьма желательной является потеря антигена e вируса гепатита B (HBeAg) у HBeAg-положительных пациентов, поскольку она отражает наличие частичного иммунного контроля над инфекцией. В качестве дополнительной цели следует рассматривать нормализацию уровня АЛТ.
Оптимальной конечной точкой лечения выступает устойчивая потеря HBsAg, так как она указывает на глубокую супрессию репликации HBV и экспрессии вирусного белка, свидетельствуя о функциональном излечении (вирусная супрессия на протяжении не менее чем 6 месяцев) хронического вирусного гепатита B, то есть когда вирус не полностью элиминирован (устранен) из организма, но иммунная система контролирует его без каких-либо лекарственных препаратов [1] [2].
Доступные медикаментозные подходы к лечению хронического вирусного гепатита B, представленные пэгинтерфероновой терапией и назначением нуклеозидных/нуклеотидных аналогов (NA), не могут похвастаться безоговорочной эффективностью. Так, потеря HBsAg происходит весьма редко: по прошествии 6 месяцев после годичного курса лечения это наблюдается в 3–7% случаев пэгинтерфероновой терапии и 0–3% случаев терапии NA. Если применение NA продолжается долго, скажем, 5–8 лет, вероятность потери HBsAg повышается, но опять же незначительно: до 10–12% у изначально HBeAg-положительных пациентов и до менее чем 1–2% у HBeAg-отрицательных [1].
Столь скромная эффективность лечения хронического вирусного гепатита B обусловлена тем, что полная эрадикация HBV нынешними препаратами затруднена по причине сохранения в гепатоцитах как ковалентно замкнутой кольцевой ДНК (кзкДНК) HBV, так и интегрированной в их ядро ДНК HBV, являющихся транскрипционными шаблонами для возобновления репликации ДНК HBV [3] [4].
Попытки комбинированного лечения обеспечили потерю HBsAg в 14% случаев, если после минимум 48-недельной NA-терапии переключить пациентов на пэгинтерфероновую терапию [5]. Считается, что прямая противовирусная активность NA, за счет ингибирования вирусной ДНК-полимеразы (обратной транскриптазы) приводящая к вирусологической супрессии и подавлению репликации HBV, частично восстанавливает адаптивный иммунитет, тем самым способствуя улучшению иммуномодулирующего действия пэгинтерферона, проявляющегося в предотвращении образования белков HBV и деплеции (истощении) внутрипеченочного пула кзкДНК [6] [7] [8] [9]. Тем не менее подход нуждается в дополнительных уточняющих исследованиях.
Помимо функционального излечения хронического вирусного гепатита B, существует куда менее достижимая цель стерильного излечения, когда HBsAg не обнаруживается, а ДНК HBV, включая кзкДНК и интегрированную, уничтожена.
В клиническом испытании B-Clear (NCT04449029) бепировирсен продемонстрировал высокую эффективность лечения, если отталкиваться от того факта, что потеря HBsAg установлена для почти трети пациентов после относительно короткого 24-недельного курса лечения.
Вирусологический ответ был зафиксирован среди как HBеAg-отрицательных пациентов, так и придерживающихся NA-терапии HBеAg-положительных. Это свидетельствует о том, что целевая для бепировирсена терапевтическая мРНК-последовательность HBV присутствует даже тогда, когда HBsAg получен из интегрированных вирусных геномов [10].
Отмеченный рост уровня АЛТ, сопутствовавший снижению уровня HBsAg, указывает на благотворные иммунные процессы. Известно, что повышение уровня АЛТ, суррогатного маркера воспаления печени, непрямым образом отражает факт иммуноопосредованного разрушения и клиренса инфицированных гепатоцитов и является предиктором потери HBsAg [11] [12].
Нельзя сказать, что нынешние результаты клинической проверки бепировирсена оказались разочаровывающими. Да, статус функционального излечения хронического вирусного гепатита B, подтвержденный отсутствием HBsAg и ДНК HBV на протяжении 6 месяцев после завершения лечения, зафиксирован у максимум 10% и 14% пациентов — соответственно среди проходивших фоновую терапию NA и без таковой. Однако с учетом короткого курса лечения эффективность следует воспринимать с должным оптимизмом.
Поскольку бепировирсен проявил наибольшую эффективность среди пациентов с изначально низким уровнем HBsAg, в дальнейшем следует рассматривать последний как основополагающий критерий выбора подходящих больных, которые с повышенной вероятностью извлекут пользу от лечения.
Кроме того, одним из предикторов успеха является статус HBeAg. В группе 1 среди HBeAg-отрицательных участников функционально излечились 10% и 14% — соответственно среди проходивших фоновую терапию NA и без таковой. При HBeAg-положительном статусе излечение отмечено для 6% и 0%.
К счастью, наблюдается тенденция, что большинство пациентов с хроническим вирусным гепатитом B являются HBeAg-отрицательными, то есть ДНК-последовательности HBV интегрированы в геном хозяина и являются основным источником HBsAg [10] [13].
Несмотря на множество изучаемых экспериментальных подходов к лечению хронического вирусного гепатита B [14], в их отношении назревает критика: мол, большинство из них фундаментально заблуждаются [15]. Инфекция HBV характеризуется очень высокой степенью генетической пластичности (тысячи квазивидов существуют у каждого отдельного пациента) [16], что является результатом отсутствия коррекционной активности обратной транскриптазы HBV, высокой скорости обновляемости (turnover) кзкДНК и постоянного иммунного давления, оказываемого на вирус. Учитывая, что даже одиночные точечные мутации HBV отменяют способность антисмысловых олигонуклеотидов (ASO) и малых интерферирующих РНК (миРНК) к специфическому расщеплению мРНК [17], под сомнение ставится даже теоретическая состоятельность данных классов лекарственных соединений для лечения хронического вирусного гепатита B.
На вышесказанное намекает тот факт, что снижение уровня HBsAg, обеспеченное бепировирсеном, оказалось сильнее среди пациентов с изначально более низким уровнем HBsAg (≤ 3000 МЕ/мл), что противоречит заявленному механизму действия препарата: сила снижения HBsAg не должна зависеть от его исходного уровня.
В предшествовавшем клиническом испытании NCT02981602 фазы IIa наблюдалась аналогичная картина [18]. Опять же, GSK3389404, вариант бепировирсена, конъюгированный с N-ацетилгалактозамином (GalNAc) в целях более таргетной доставки в гепатоциты [19], не оказал существенного влияния на снижение уровня HBsAg [20].
«Кавигейл» (Kavigale, сипавибарт) — новый лекарственный препарат, предназначенный для доконтактной профилактики (PrEP) коронавирусной инфекции COVID-19, вызванной коронавирусом SARS-CoV-2, у взрослых и подростков (12 лет и старше, весом как минимум 40 кг) с ослабленным иммунитетом по состоянию здоровья или ввиду приема иммуносупрессивных препаратов.
ОСНОВНЫЕ ФАКТЫ
«Кавигейл» снизит риск развития симптоматического ковида, вызванного любым вариантом коронавируса, а также риск развития осложнений инфекции, требующих госпитализации.
По сути «Кавигейл» представляет собой альтернативу стандартной противоковидной вакцине. Но применяться он должен только в том случае, если человек по каким-либо причинам, связанным со здоровьем или нынешним лечением, не может привиться от коронавируса.
«Кавигейл» вводится внутримышечной инъекцией один раз в полгода.
Сипавибарт (sipavibart) — противовирусное моноклональное антитело, наделяющее пассивной иммунизацией против SARS-CoV-2 путем блокирования проникновения коронавируса в клетки: вирус теряет возможность для своего размножения, в результате становясь неинфекционным.
«Кавигейл» предложен «АстраЗенека» (AstraZeneca) взамен ушедшего в прошлое «Эвушелда» (Evusheld, тиксагевимаб + цилгавимаб), который был первым препаратом, предназначенным для противоковидной PrEP-защиты, но который весьма быстро лишился своей эффективности ввиду появления новых омикрон-вариантов SARS-CoV-2.
В середине декабря 2024 года Комитет по лекарственным препаратам для медицинского применения (CHMP) при Европейском агентстве по лекарственным средствам (EMA) отрекомендовал одобрить «Кавигейл». Решение регулятора не за горами [1].
В США и других странах сипавибарт, если будет официально разрешен, может получить иное брендовое название: например, «Нексшелд» (Nexsheld), «Нексшелда» (Nexshelda), «Райшелда» (Ryshelda).
Пемивибарт каждые три месяца — для тех, кто не может привиться против COVID-19.
ПОЧЕМУ ЭТО ВАЖНО
По данным реальной клинической практики, люди с ослабленной иммунной системой испытывают непропорционально большое бремя коронавирусной инфекции COVID-19 по сравнению с общей популяцией, гораздо чаще сталкиваясь с тяжелым течением вызванного коронавирусом SARS-CoV-2 заболевания и его неблагоприятными последствиями.
Так, согласно обсервационному популяционному исследованию INFORM, охватившему резидентов Великобритании, люди с ослабленным иммунитетом подвержены существенно большим рискам госпитализации или смерти по причине осложнений ковида: для определенных категорий эти риски вырастают в 13 и 20 раз. Несмотря на то что таких индивидуумов насчитывается лишь 3,9% населения, они вносят весомый вклад в статистику неблагополучных ковидных исходов: в 2022 году на их долю пришлось 22% случаев госпитализации, 28% случаев поступления в отделение интенсивной терапии, 24% смертельных исходов. И всё это не взирая на то, что 84% людей с ослабленным иммунитетом получили как минимум три дозы противоковидных вакцин [1].
Согласно обсервационному когортному исследованию EPOCH-US среди резидентов США, проведенному в период с апреля 2020 года по март 2022-го, люди с ослабленным иммунитетом, которых насчитывается 2,7% населения, требуют выделения более чем вчетверо больших ресурсов здравоохранения, чем представители общей популяции, при госпитализации с коронавирусной инфекцией COVID-19 [2].
КЛИНИЧЕСКАЯ ПРОВЕРКА
Клиническое исследование SUPERNOVA (NCT05648110) фазы III (рандомизированное, двойное слепое, плацебо-контролируемое и с активным контролем, многоцентровое, международное) пригласило добровольцев (n=3335) в возрасте 12 лет и старше в целях изучения эффективности и безопасности «Кавигейла» /«Нексшелда» (Kavigale / Nexsheld, сипавибарт) в сравнении с «Эвушелдом» (Evusheld, тиксагевимаб + цилгавимаб) в задаче доконтактной профилактики коронавирусной инфекции COVID-19.
Среди основных требований к участникам: отрицательный экспресс-тест на антиген COVID-19 и наличие статуса ослабленного иммунитета по такой причине, как солидная или гематологическая злокачественная опухоль, трансплантированный солидный орган или гемопоэтические стволовые клетки, прием иммуносупрессивных препаратов, лечение CAR-T-клетками или истощающая пул B-клеток терапия, гемодиализ, умеренный или тяжелый первичный иммунодефицит.
Участникам назначали внутримышечные инъекции: либо 300 мг сипавибарта (sipavibart), либо 300 мг тиксагевимаба (tixagevimab) с 300 мг цилгавимаба (cilgavimab) — дважды с интервалом в 6 месяцев между дозами.
В этот годичный период проводилась сравнительная оценка нейтрализующей активности «Кавигейла» /«Нексшелда» и «Эвушелда», а также их эффективности в предотвращении развития симптоматической коронавирусной инфекции COVID-19, вызванной любым вариантом коронавируса SARS-CoV-2 и его вариантами без мутации F456L. Собирались также данные, касающиеся случаев симптоматического ковида, заболевания в тяжелой форме, госпитализации по по причине осложнений, смертельных исходов.
Исследование, начавшееся в середине декабря 2022 года, должно было завершиться в конце марта 2023-го. Осталось дождаться публикации полных его результатов.
«АстраЗенека» также осуществила клиническое исследование NOVELLA (NCT06057064) фазы II, проверившее «Нексшелд» среди жителей России (n=116).
ЭФФЕКТИВНОСТЬ
В ходе разработки сипавибарта (sipavibart) преследовалась следующая задача: сделать это моноклональное антитело таким образом, чтобы его нейтрализующая активность оставалась максимально высокой против, во-первых, прежде доминировавших вариантов (разновидностей) коронавируса SARS-CoV-2 и, во-вторых, тех его новых вариантов, которые становятся всё более распространенными в настоящее время.
И если первая часть этой задача вполне себе реализуема, то со второй неизбежно возникают сложности, поскольку SARS-CoV-2 проходит безостановочный процесс эволюционных генетических изменений, потенциально способных снизить эффективность любого профилактического или лекарственного препарат либо вообще превратить его в неработающий.
Вот почему нельзя безоговорочно заявлять, что сипавибарту по силам обеспечить абсолютную защиту от коронавирусной инфекции COVID-19. Тем не менее некоторые достоверные предположения сделать всё же можно — даже без наличия результатов клинической проверки препарата «Кавигейл» /«Нексшелд» (Kavigale / Nexsheld, сипавибарт).
Так, согласно данным in vitro сипавибарт сохраняет нейтрализующую активность против всех исторических вариантов SASR-CoV-2, а также омикрон-подвариантов, включая JN.1, BA.2.86 и XBB.1.16. Однако он ее теряет в случае омикрон-подвариантов с мутацией F456L, таких, например, как XBB.1.5.10 / EG.5, EG.5.1, KP.1.1, LB.1, KP.3.3 [1]. Пока нет данных, касающихся нейтрализующей активности сипавибарта против набирающих силу F456L-мутантных разновидностей KP.3.1.1 и XEC.
Согласно результатам клинической проверки SUPERNOVA (NCT05648110) сипавибарт в целом снизил риск развития симптоматической коронавирусной инфекции COVID-19 на 30% относительно «Эвушелда» (Evusheld, тиксагевимаб + цилгавимаб). Если отбросить варианты коронавируса с F456L-мутацией, снижение составило относительных 35% [2].
Европейское агентство по лекарственным средствам (EMA) настоятельно не рекомендует использовать сипавибарт в тех странах и регионах, где, согласно текущей эпидемиологической обстановке, преобладают F456L-мутантные варианты SARS-CoV-2: препарат, скорее всего, не принесет никакой клинической пользы. В таком случае единственно верным решением защиты от ковида остается вакцинация — даже лиц с ослабленной иммунной системой.
Для более точного понимания потенциальной степени защитной эффективности препарата «Кавигейл» /«Нексшелд» и принятия информированного решения на предмет оправданности его профилактического назначения, следует изучить следующие наборы данных:
Мониторинг Всемирной организации здравоохранения (ВОЗ) циркулирующих в настоящее время вариантов SARS-CoV-2, вызывающих интерес (ВВИ), и вариантов SARS-CoV-2 под наблюдением (ВПН) [3].
Распространенность того или иного варианта SARS-CoV-2 в конкретном регионе или стране [4] [5] [6] [7] [8].
Базы данных, аккумулирующие сведения о снижении чувствительности вариантов SARS-CoV-2 к нейтрализующей активности моноклональных антител [9].
Релевантные научные, исследовательские и презентационные публикации [10] [11].
Инструкция по медицинскому применению препарата «Кавигейл» /«Нексшелд» (Kavigale / Nexsheld, сипавибарт), в которой должны быть указаны подробные сведения, касающиеся нейтрализации вариантов SARS-CoV-2 с информацией о снижении чувствительности и развитии резистентности.
ВОПРОС ЦЕНЫ
Цена препарата «Кавигейл» /«Нексшелд» (Kavigale / Nexsheld, сипавибарт) будет объявлена, когда он получит регуляторное одобрение.
Можно смело полагать, что стоимость сипавибарта (sipavibart) никак не будет выставлена ниже 2 тыс. долларов. Это типичная цена для моноклональных антител против коронавируса SARS-CoV-2, когда они еще эффективно работали и были повсеместно разрешены для применения [1] [2] [3] [4] [5].
Для бизнеса «АстраЗенека» запуск сипавибарта определенно выгоден, если отталкиваться от заработков ушедшего в прошлое «Эвушелда», который принес 85 млн, 2,19 млрд и 312 млн долларов — соответственно в 2021, 2022 и 2023 гг.
КАК ЭТО РАБОТАЕТ
Сипавибарт (sipavibart, AZD3152) — полностью человеческое моноклональное IgG1-антитело, таргетированное на высококонсервативный эпитоп рецептор-связывающего домена (RBD) S-белка коронавируса SARS-CoV-2.
После связывания сипавибарта с RBD блокируется взаимодействие последнего с ангиотензинпревращающим ферментом 2 (ACE2) — рецептором, который коронавирус использует для прикрепления к мембране клеток организма-хозяина с последующим проникновением в них.
Пассивная иммунизация против ковида при помощи сипавибарта, предполагающая нейтрализацию SARS-CoV-2, который теряет возможность для репликации, обеспечивает временную защиту от заражения, а если инфекция всё же дала о себе знать, то следует ожидать замедления прогрессирования заболевания и ускорения выздоровления.
Сипавибарт является производным антител, полученных из B-клеток памяти выздоровевшего пациента, вакцинированного, но всё равно столкнувшегося с заражением коронавирусной инфекцией COVID-19, вызванной омикрон-вариантом BA.1 коронавируса SARS-CoV-2.
В антительный каркас сипавибарта внедрены аминокислотные замены, призванные улучшить его фармакокинетические и биохимические свойства [1]. Так, замена YTE повысила сродство антитела к неонатальному Fc-рецептору, что привело к улучшению рециркуляции и существенному продлению периода полувыведения так, что продолжительность действия препарата увеличилась более чем втрое по сравнению с обычными неоптимизированными антителами [2] [3] [4].
Продолжительное персистирование сипавибарта в организме позволяет надеяться, что противоковидная защита сохранится на протяжении не менее чем шести месяцев после однократного введения препарата [5]. Впрочем, учитывая агрессивную мутационную природу SARS-CoV-2, сипавибарт, не исключено, придется применять чаще, если поставлена задача максимизации защиты от ковида.
Аминокислотная замена TM снизила силу связывания с FcγR и C1q, что отразилось резким ослаблением или даже отсутствием эффекторной Fc-функции, включающей антителозависимый клеточный фагоцитоз (ADCP), антителозависимую клеточную цитотоксичность (ADCC), антителозависимое осаждение комплемента (ADCD), антителозависимую активацию естественных клеток-киллеров (ADNKA) и влекущей за собой иммунопатологические проявления. Минимизирован потенциальный риск антителозависимого усиления (ADE) — явления, при котором неоптимальные вирусоспецифические антитела, напротив, способствуют, а не подавляют инфекцию или заболевание [6].
PERICULUM IN MORA: ОПАСНОСТЬ В ПРОМЕДЛЕНИИ
В начале декабря 2021 года свет увидел «Эвушелд» (Evusheld, тиксагевимаб + цилгавимаб) — комбинация из моноклональных антител авторства «АстраЗенека» (AstraZeneca), ставшая первым (и единственным) способом доконтактной профилактики коронавирусной инфекции COVID-19 для тех, кому не подходит или противопоказана вакцинация.
Появление «Эвушелда» оказался настолько значимым событием, что журнал «Тайм» (Time) включил его в список лучших изобретений 2022 года [1] [2].
Однако уже в конце января 2023-го FDA отменило регистрацию «Эвушелда»: ввиду засилья новых омикрон-вариантов коронавируса SARS-CoV-2, с которыми он справиться не в силах, подобный способ защиты от инфекции работать перестал [3].
Одна доза моноклонального коктейля AstraZeneca защитит от ковида на протяжении полугода и дольше.
Другие фармразработчики так и не смогли предложить альтернативу «Эвушелду», что оставило ни с чем десятки миллионов людей, отчаянно нуждающихся в подобной пассивной иммунизации от ковида — вместо активной иммунизации, реализуемой посредством вакцинации.
«АстраЗенека», очевидно прогнозировавшая печальную участь «Эвушелда», заблаговременно подсуетилась, в середине мая 2022 года лицензировав сипавибарт (sipavibart) и другие противоковидные моноклональные антитела у британской «Ар-кью байо» (RQ Bio) и взамен пообещав до 157 млн долларов плюс роялти от реализации готового препарата [4].
Клиническая проверка сипавибарта началась в середине декабря 2022 года [5].
«АстраЗенека» рассчитывала сделать сипавибарт доступным для широкого применения во второй половине 2023 года [6].
Для быстрого вывода «Кавигейла» / «Нексшелда» (Kavigale / Nexsheld, сипавибарт) на открытый рынок существовали абсолютно все условия, ведь препарат взял на вооружение точно такой же глубоко оптимизированный каркас антитела, на базе которого построен «Эвушелд», то есть никаких конструктивных доработок не требовалось. Фактически стояла единственная задача: оценить защитную эффективность сипавибарта в противодействии новым вариантам коронавируса SARS-CoV-2, которые перестали реагировать на «Эвушелд».
Но не сложилось.
В середине марта 2024 года «Инвивид» (Invivyd) выпустила «Пемгарда» (Pemgarda, пемивибарт) — моноклональное антитело, предназначенное для доконтактной профилактики (PrEP) коронавирусной инфекции COVID-19, вызванной коронавирусом SARS-CoV-2, у взрослых и подростков.
Пемивибарт каждые три месяца — для тех, кто не может привиться против COVID-19.
ОБРАТНАЯ СТОРОНА
В отличие от США [1] на многих территориях, включая Россию, Европу, Великобританию, Канаду, Австралию и пр., «Эвушелд» (Evusheld, тиксагевимаб + цилгавимаб) и/или другие противоковидные моноклональные антитела и их комбинации по-прежнему включены в клинические рекомендации по профилактике и/или лечению коронавирусной инфекции COVID-19 [2] [3] [4] [5] [6].
Что примечательно, сами регуляторы давно выпустили многочисленные предупреждения о недостаточной эффективности моноклональных антител в борьбе с новыми вариантами SARS-CoV-2 в эпоху омикрон-линии, но до сих пор не отменили их маркетинговые авторизации [7] [8] [9] [10]. Не исключено, отраслевые контролеры заняли выжидательную позицию: вдруг коронавирус мутирует так, что моноклональные антитела вновь смогут с ним сражаться?
Авторитетные медицинские агентства высказываются резко против любых противоковидных моноклональных антител по причине полного отсутствия у них должной эффективности: коронавирус SARS-CoV-2 слишком глубоко и сильно мутировал, чтобы эти препараты, прежде работавшие, продолжали обеспечивать необходимую противовирусную нейтрализующую активность [11] [12] [13].
Согласно обновляемой базе данных Стэнфордского университета, отслеживающей снижение чувствительности вариантов коронавируса к нейтрализующей активности моноклональных антител, таргетированных на S-белок SARS-CoV-2, ситуация удручает: препараты этого класса в целом не пригодны к войне против ковида с его нынешним набором мутаций [14].
Кроме «Пемгарда» (Pemgarda, пемивибарт) — да и то уже с большой натяжкой! — никакие из когда-либо официально одобренных противоковидных моноклональных антител, пусть даже ранее относительно успешно применявшихся в профилактике коронавирусной инфекции COVID-19 (доконтактной и постконтактной) и ее лечении, сейчас использовать нет никакого смысла. Среди таких препаратов, в настоящее время абсолютно бесполезных [15] [16] [17] [18] [19] [20] [21] [22] [23] [24]:
Позиция доказательной медицины проста и понятна: зачем платить большие деньги за абсолютно никчемные препараты и при этом тщетно обнадеживать людей, будто бы они верно защищены от ковида?