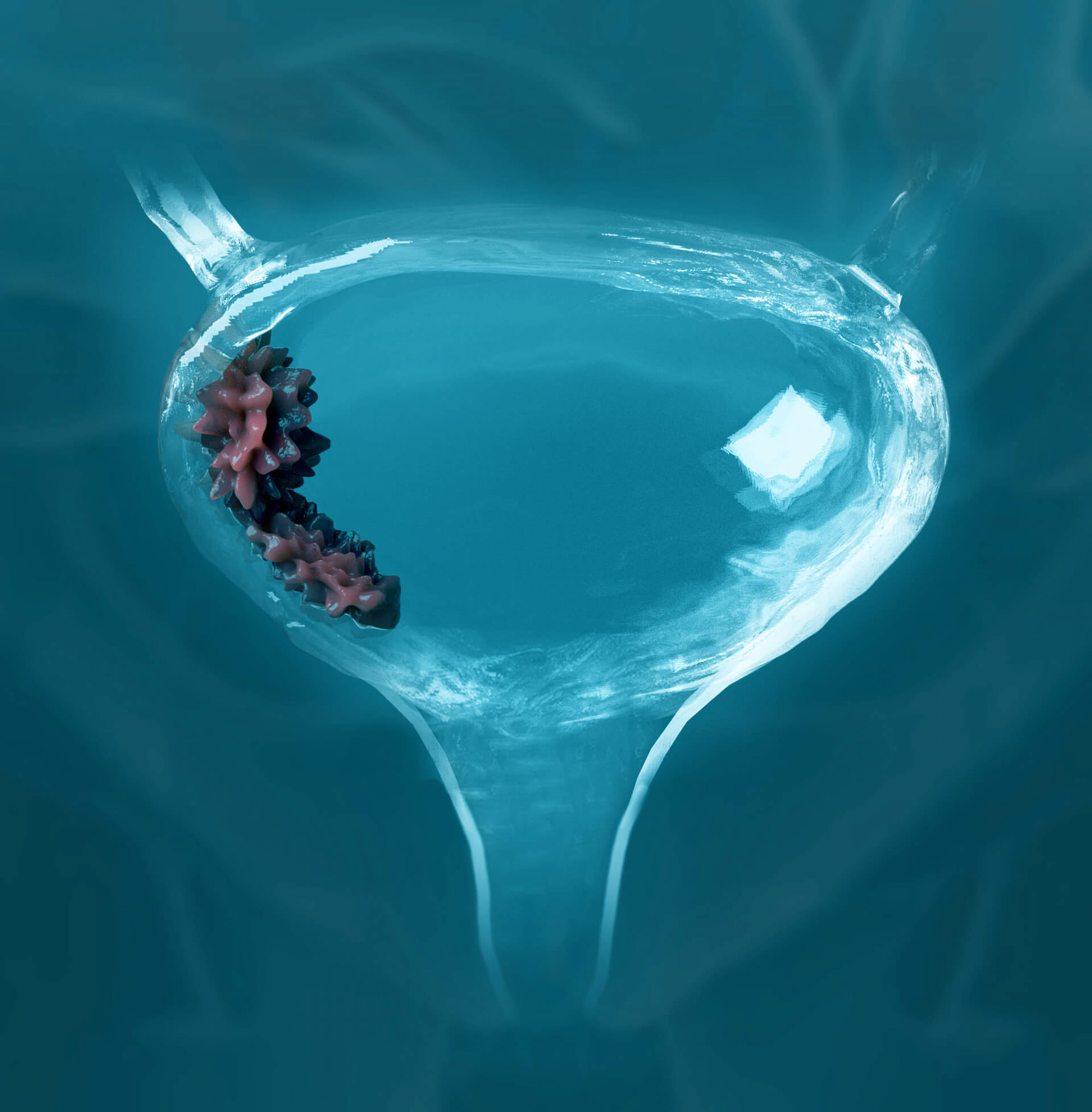
«Гилеад сайенсиз» (Gilead Sciences) замыслила сделать антиретровирусную терапию (АРТ) инфекции вируса иммунодефицита человека 1 (ВИЧ-1) намного более удобной, простой и практичной для людей, живущих с этим заболеванием.
ОСНОВНЫЕ ФАКТЫ
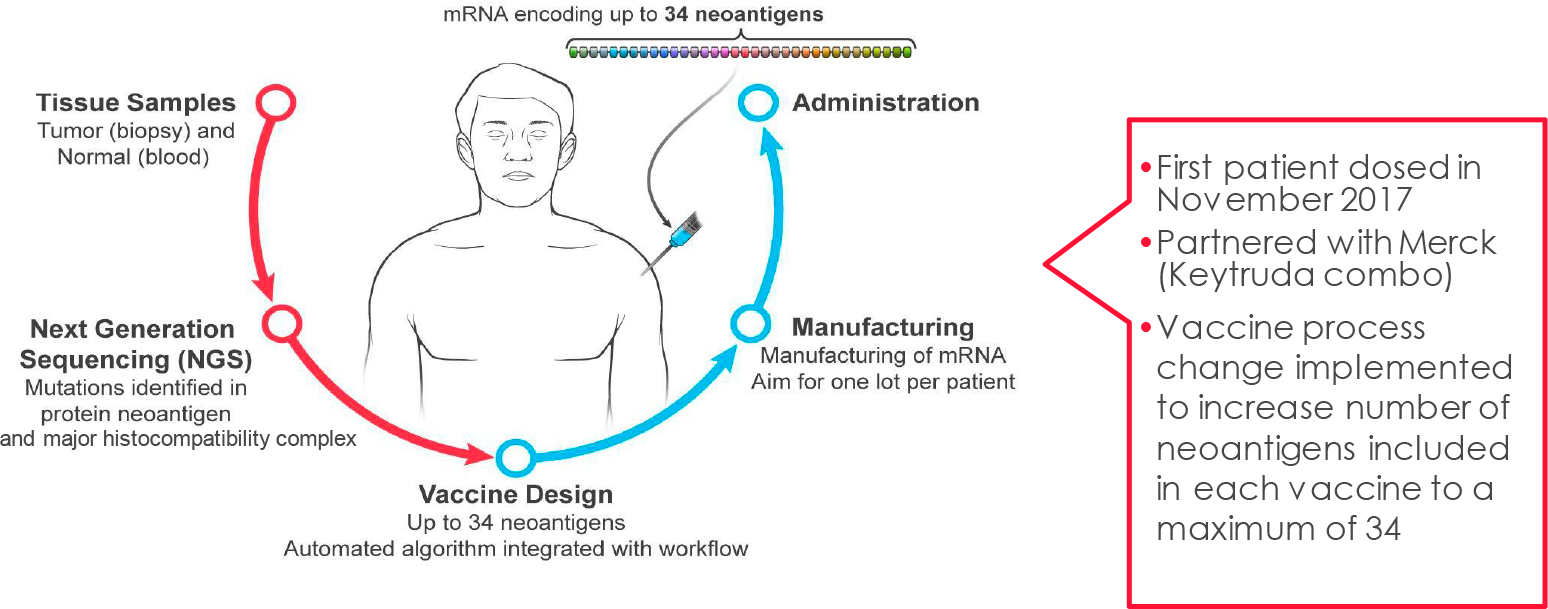
Продолжается клиническая разработка комбинации из трех лекарственных препаратов: ленакапавира (lenacapavir; LEN), теропавимаба (teropavimab; TAB) и зинлирвимаба (zinlirvimab; ZAB).
Ленакапавир (LEN), первый представитель АРТ-класса ингибиторов капсида ВИЧ, уже одобрен под брендом «Санленка» (Sunlenca) для лечения этой инфекции с мультилекарственной устойчивостью (МЛУ).
Экспериментальные теропавимаб (GS-5423, 3BNC117) и зинлирвимаб (GS-2872, 10-1074) — нейтрализующие антитела широкого спектра действия (bNAb), которые «Гилеад» лицензировала у Рокфеллеровского университета в январе 2020 года [1]. Эти антитела, выделенные у лиц с сильной иммунной реакцией в ответ на ВИЧ, оказывают как прямой противовирусный эффект, так и иммуноопосредованный, инициирующий запуск иммунных сигнальных схем организма.
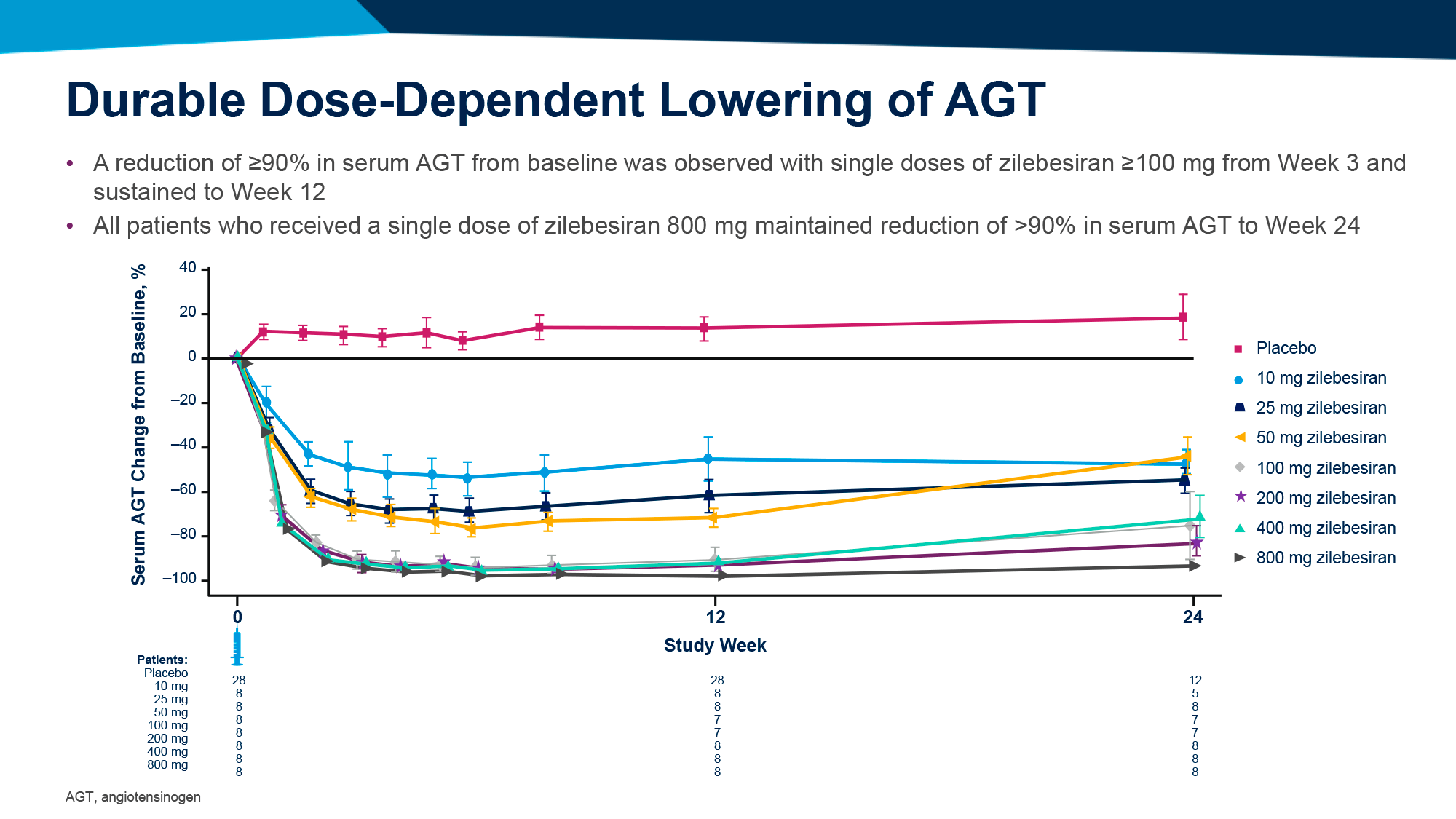
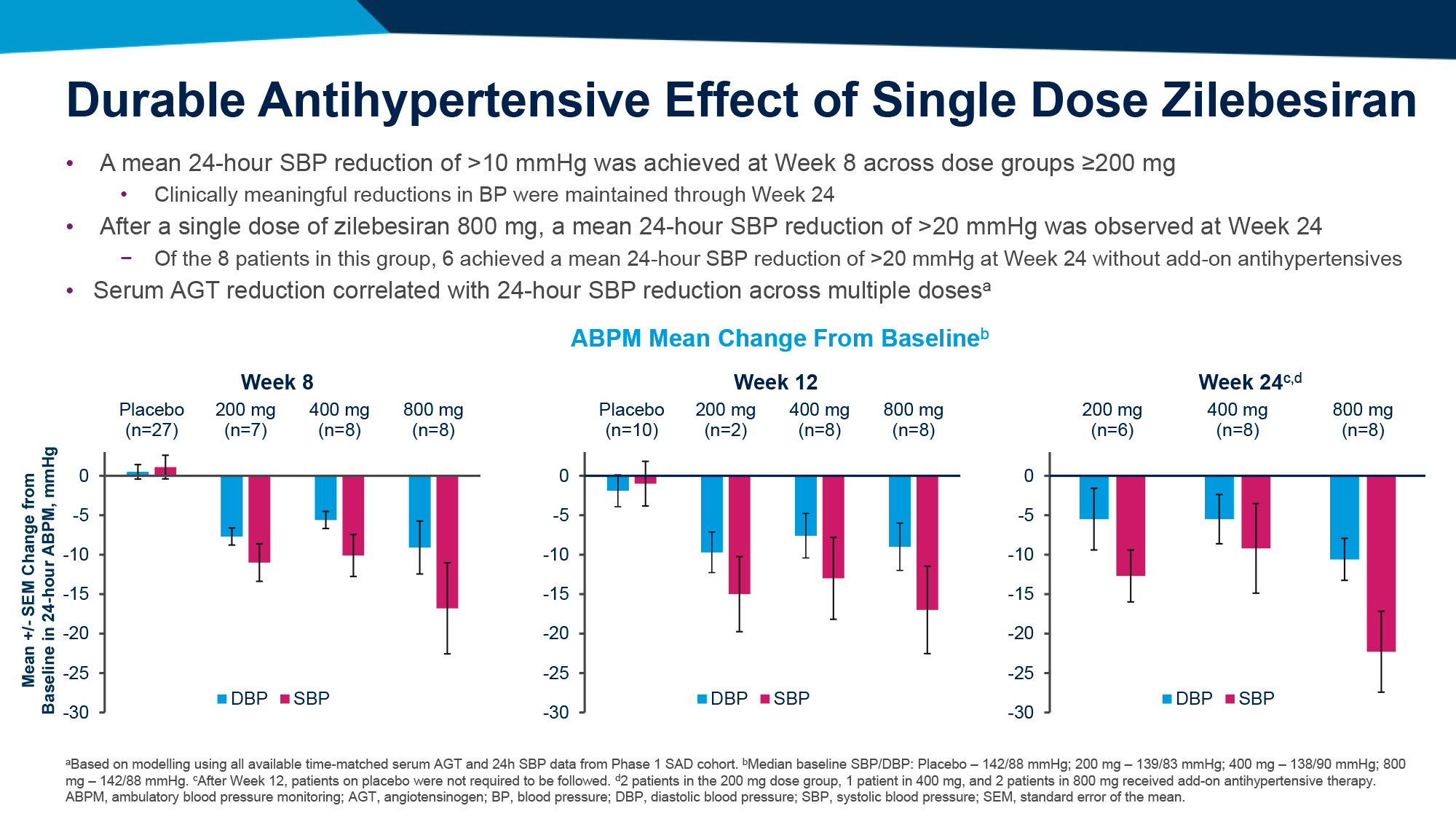
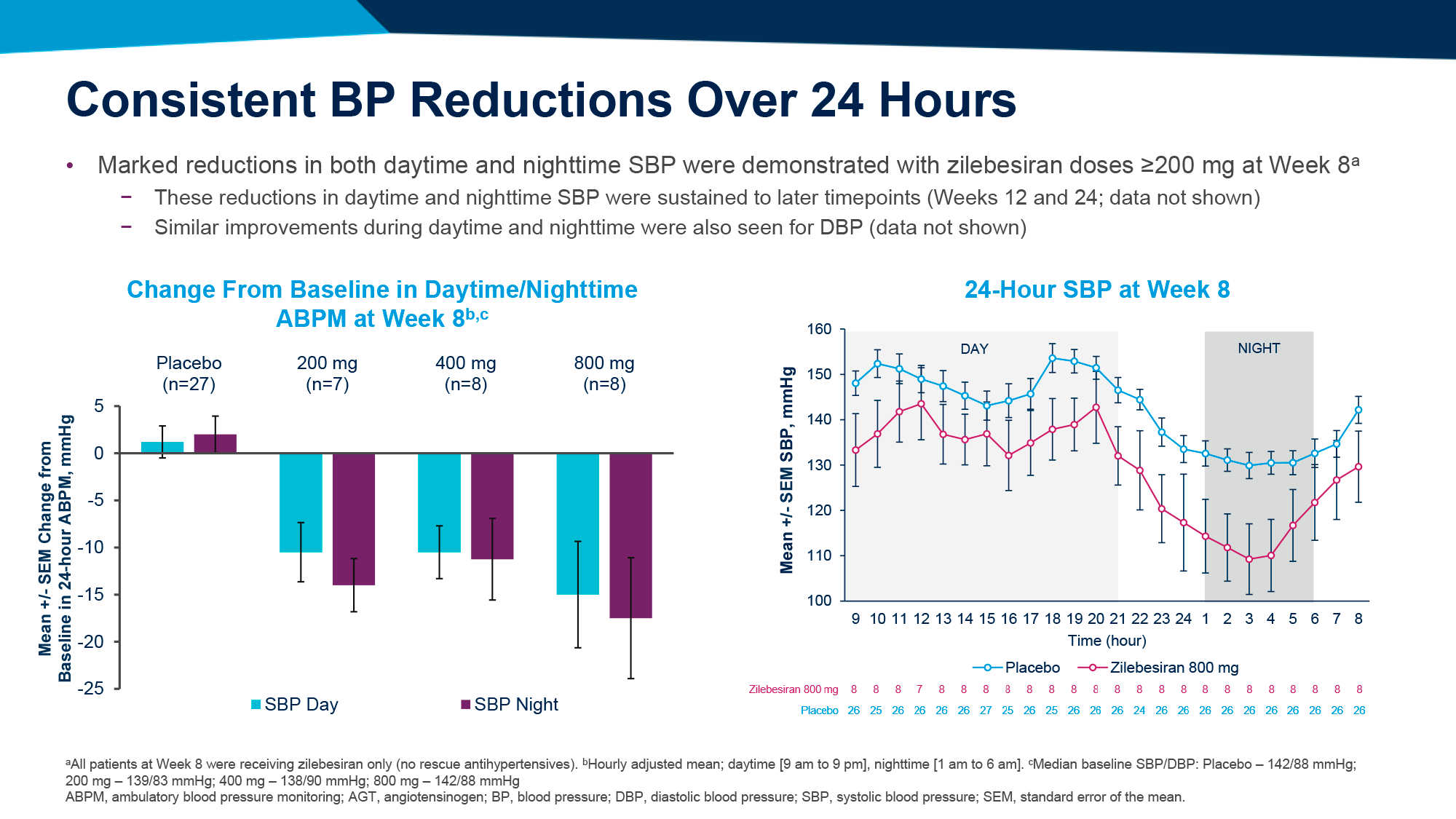
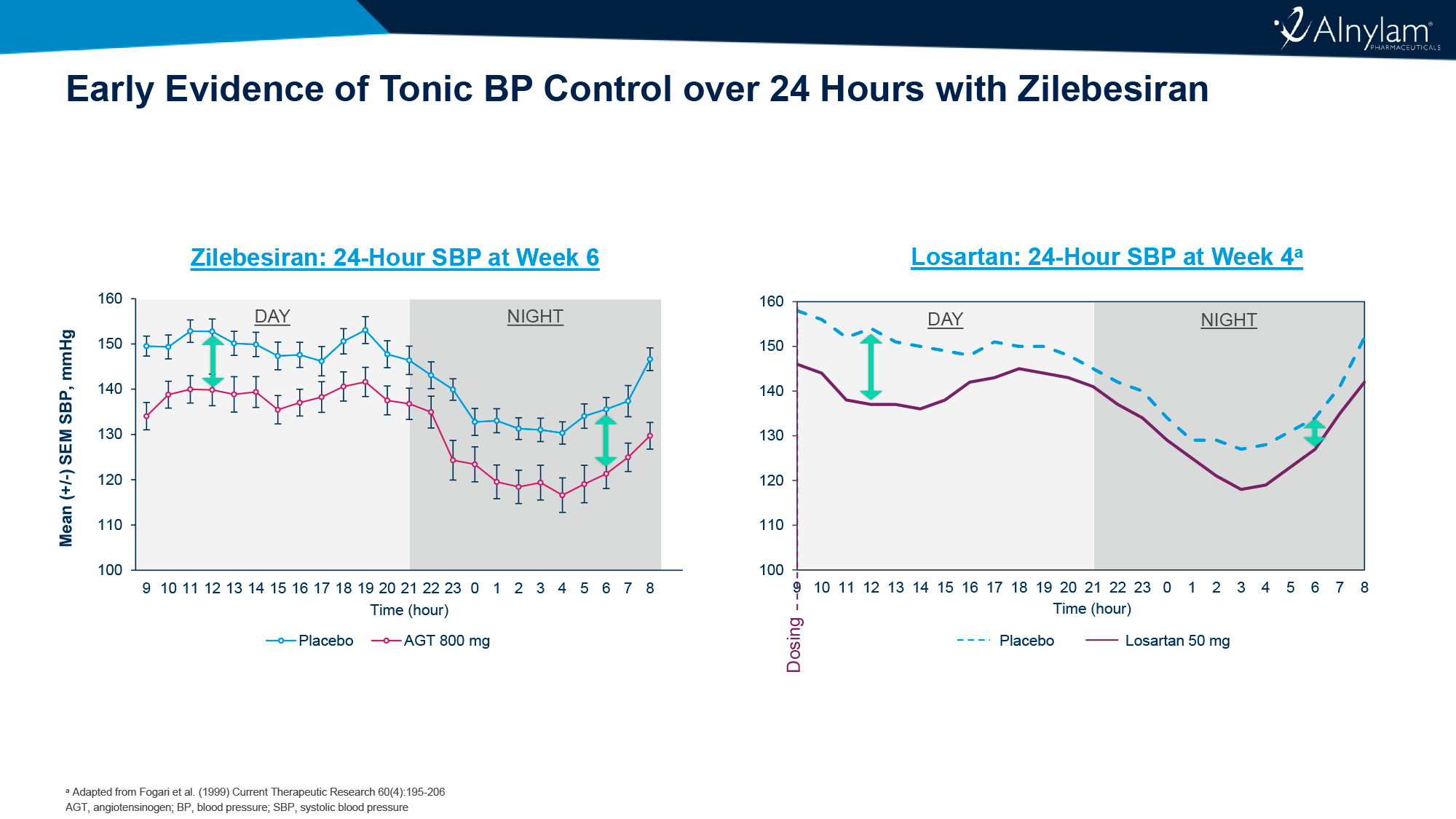
После однократного введения подкожного LEN и внутривенных TAB и ZAB и последующих наблюдений на протяжении 26 недель вирусная супрессия была подтверждена для 87% испытуемых (n=26/30), которые до исследования уже находились в этот статусе.
У тех, у кого случилась вирусная отдача (n=3), вирусная нагрузка оставалась приемлемо низкой.
С лекарственной резистентностью к ленакапавиру столкнулся 1 человек, к bNAb — никто.
Ленакапавир примкнул к ибализумабу и фостемсавиру в борьбе с резистентным вирусом иммунодефицита человека.
ПРЯМАЯ РЕЧЬ
«Использование собственной иммунной системы организма для борьбы с ВИЧ должно улучшить общие результаты лечения».
Дайана Брейнард (Diana Brainard), старший вице-президент по ВИЧ и новым вирусам «Гилеад сайенсиз» (Gilead Sciences).
«Наши ученые собрали обширный материал, подтверждающий, что нейтрализующие антитела способны коренным образом изменить парадигму лечения инфекции ВИЧ».
Жанна Фаррелл (Jeanne Farrell), помощник вице-президента по развитию технологий Рокфеллеровского университета (США).
«Как же далеко продвинулось лечение ВИЧ-инфекции! Если в самом начале пациентам приходилось просыпаться каждые 4 часа, чтобы принять лекарства, — вплоть до 15–20 таблеток ежедневно, то сейчас мы можем вкалывать АРТ-препараты через месяц. Теперь открылась перспектива лечиться всего два раза в год».
Джозеф Дж. Эрон (Joseph J. Eron), директор Центра исследований СПИДа при Университете Северной Каролины (США).
СУТЬ ВОПРОСА
Появление мощных, удобных, безопасных и с хорошей переносимостьх однотаблеточных схем комбинированной АРТ произвело революцию в оказании помощи людям, живущим с ВИЧ. Глобальное внедрение таких схем привело к значительному снижению распространения синдрома приобретенного иммунодефицита (СПИД) и смертности от его осложнений во всём мире [1].
Если лечение начато сразу после постановки диагноза, можно рассчитывать на нормальную или близкую к нормальной продолжительность жизни [2]. Так, например, внедрение АРТ в ЮАР увеличило продолжительность жизни более чем на десять лет [3]. Более того, применение эффективной АРТ резко снижает риск передачи ВИЧ половым партнерам [4].
Состоятельность этих впечатляющих результатов напрямую зависит от постоянной приверженности ежедневной пероральной терапии на протяжении всей жизни. Прерывание АРТ приводит к вирусному рецидиву в среднем через 10–14 дней; и через 28 дней почти у всех больных [5]. Недостаточная приверженность несет риск появления лекарственно-устойчивых штаммов вируса, которые плохо реагируют на АРТ-препарат и снижают вероятность успешного ответа на последующие АРТ-схемы.
Необходимость поддерживать стабильно высокий уровень приверженности АРТ представляет собой серьезную проблему для многих людей, живущих с ВИЧ: особенно для тех, кто страдает психическими заболеваниями, употребляет психоактивные вещества и испытывает иные психосоциальные стрессы. Кроме того, многие пациенты с превосходной приверженностью оказываются фактически запертыми в ситуации, когда ежедневный прием пероральных АРТ-препаратов является нежелательным напоминанием об их ВИЧ-статусе и связанной с ним стигмой. По этим причинам разработка АРТ-схем длительного действия является важным приоритетом в данной области.
Инъекционное сочетание каботегравира (cabotegravir; CAB), ингибитора переноса цепи интегразой (INSTI), и рилпивирина (rilpivirine; RPV), ненуклеозидного ингибитора обратной транскриптазы (NNRTI), реализованное под американским брендом «Кабенува» (Cabenuva) и европейскими «Вокабриа» (Vocabria) и «Рекамбис» (Rekambys), — первая АРТ-схема длительного действия, вводимая ежемесячно или каждые два месяца и одобренная для лечения ВИЧ у людей в статусе вирусной супрессии. Клиническая проверка подтвердила, что комбинация не хуже, чем пероральный АРТ-режим [6] [7].
«Кабенува» / «Вокабриа» + «Рекамбис» не хуже, чем «Биктарви».
Впоследствии, однако, выяснилось, что у 4 из 6 участников с подтвержденной вирусологической неудачей появились мутации, ассоциированные с резистентностью к CAB [8]. Вот почему перспективы этой схемы лечения сдерживаются опасениями по поводу возможного возникновения резистентности к INSTI, которые составляют основу АРТ-схем первой линии во всём мире. К исходным факторам, связанным с повышенным риском подтвержденной вирусологической неудачи при назначении «Кабенувы», относятся индекс массы тела (ИМТ) ≥ 30 кг/м2, наличие ВИЧ-1 подтипа A6 или A1 (одни из наиболее распространенных в России и странах СНГ) и мутаций резистентности к RPV [9].
Таким образом, по-прежнему не закрыт вопрос с разработкой и внедрением более совершенных и надежных АРТ-схем длительного действия.
БОЛЬШИЕ ЧИСЛА
Статистические сведения образца 2023 года следующие [1] [2]:
Во всём мире насчитывается 39,9 млн людей, живущих с ВИЧ.
Если сейчас у 77% (30,7 млн) инфицированных ВИЧ есть доступ к АРТ, то в 2000 году эффективно лечиться могли менее 2% (!).
С начала эпидемии ВИЧ им заразились 88,4 млн, а от осложнений СПИДа умерла почти половина больных — 48%, или 42,3 млн человек.
Число новых случаев ВИЧ-инфицирования снизилось на 60% с пикового показателя заболеваемости в 1995 году: в 2023 году диагноз был впервые поставлен 1,3 млн человек — против 3,3 млн в 1995-м.
ВИЧ-инфекция справедлива главным образом для взрослых: 97%, или 38,6 млн зараженных.
Количество смертей от осложнений СПИДа снизилось на 69% (630 тыс.) с пиковых 2,1 млн в 2004 году. Тем не менее всё равно кто-то умирает буквально каждую минуту.
Маргинализация, дискриминация и в некоторых случаях криминализация приводят к повышенной распространенности ВИЧ среди определенных групп населения (в порядке убывания риска): трансгендеров, мужчин, практикующих секс с мужчинами (МСМ), потребителей инъекционных наркотиков, секс-работников, молодых женщин и девушек из некоторых стран Африки, заключенных.
КАК ЭТО РАБОТАЕТ
Поскольку эффективной вакцины против ВИЧ сейчас нет и в ближайшем будущем не предвидится, необходимы новые подходы к профилактике передачи вируса. Такие стратегии включают использование антител широкого спектра действия (bNAb), выделенных от инфицированных людей с высоким титром нейтрализующей активности против ВИЧ-1 [1] [2] [3]. Фактически речь идет о пассивной иммунизации против этого вируса.
Этим антителам под силу нейтрализовать большинство циркулирующих штаммов вируса, нацеливаясь на различные неперекрывающиеся эпитопы на шипе оболочки ВИЧ-1, такие как CD4-связывающий сайт [3] [4] [5], вариабельная петля 1 и 2 (петля V1V2) [2] [6], вариабельная петля V3 [1] [7] [8], проксимальная область мембраны [9] и ряд эпитопов, охватывающих взаимодействующую область gp120–gp41 [10] [11].
Несколько bNAb, в том числе 3BNC117, VRC01, PGT121 и 10-1074, способны защищать макак от инфекций, вызванных вирусом иммунодефицита обезьян (SHIV) [12] [13] [14] [15] [16] [17]. Антитела также контролируют репликацию вируса у обезьян, хронически инфицированных SHIV [18] [19] [20] [21].
Исследования на людях с использованием моноклональных антител VRC01 или 3BNC117, которые нацелены на CD4-связывающий сайт, или моноклонального антитела 10-1074, которое связывается с основанием петли V3 и окружающими гликанами, показали, что они в целом безопасны и активны in vivo [22] [23] [24] [25].
Введение bNAb транзиторно снижает виремию в плазме и задерживает рецидив при прерывании лечения у лиц с инфекцией ВИЧ-1 [22] [23] [24] [25] [26] [27].
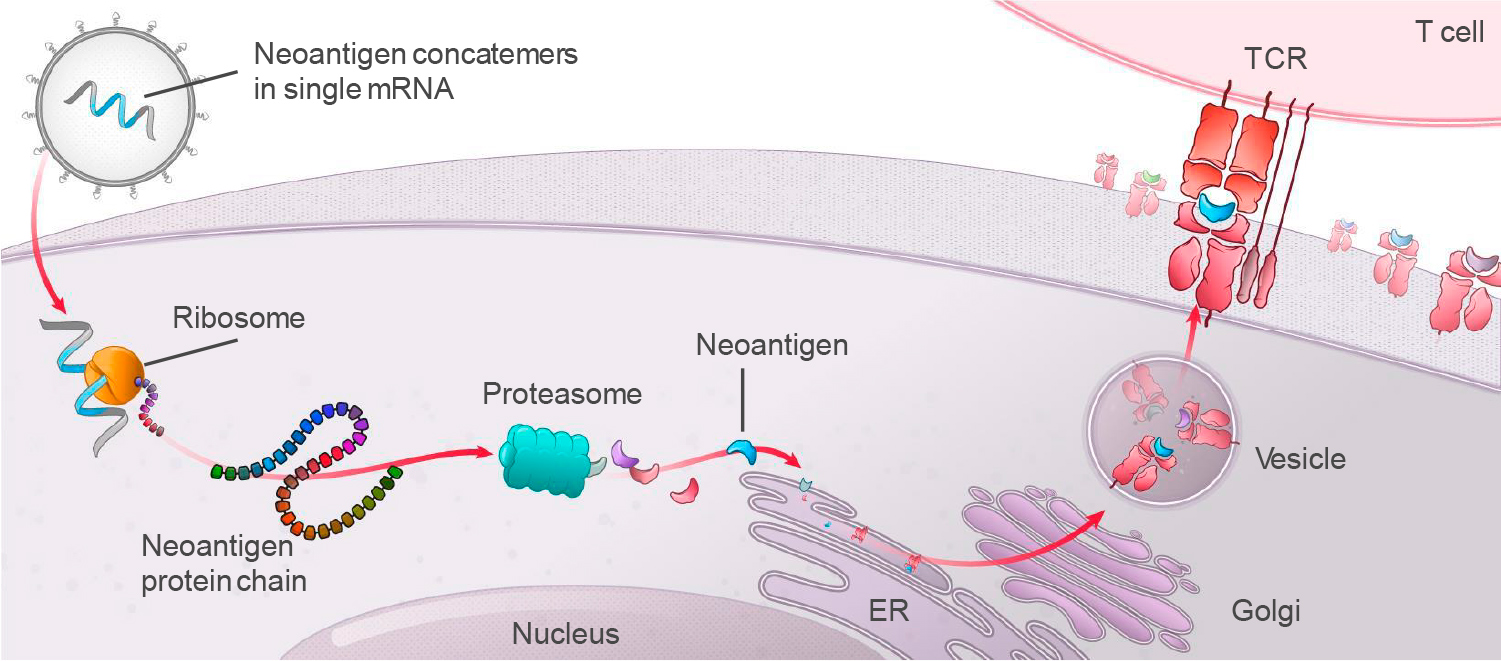
Полностью человеческие моноклональные антитела теропавимаб и зинлирвимаб связываются с оболочечным гликопротеином gp120 ВИЧ-1 (отвечает за проникновение вируса в клетку), и делают это в его разных неперекрывающихся эпитопах. Теропавимаб связывается с CD4-связывающим сайтом gp120, тогда как зинлирвимаб — с вариабельной петлей 3 (V3), которая участвует в связывании gp120 с хемокиновыми рецепторами [28]. Антитела механистически прерывают важный этап ВИЧ-инфицирования — вход вирионов в CD4+-клетки-хозяина.
[su_spoiler title=»Как ВИЧ проникает в клетку?» class=»my-custom-spoiler»]
На первом этапе слияния происходит высоаффинное прикрепление CD4-связывающих доменов gp120, оболочечного белка вируса, к трансмембранному гликопротеину CD4 клетки-хозяина. После этого конформация gp120 претерпевает структурные изменения, экспонируя (выставляя и обнажая) домены связывания хемокиновых рецепторов gp120. Эти домены связываются с корецепторами, экспрессирующими на клетке-хозяина (обычно это хемокиновые рецепторы CCR5 или CXCR4). Сформировавшееся стабильное двухточечное прикрепление позволяет gp41, вирусному N-концевому пептиду слияния, проникнуть через клеточную мембрану. Повторяющиеся последовательности в gp41, HR1 и HR2, взаимодействуют, вызывая коллапс (сворачивание) внеклеточной части gp41 в шпилькообразную форму. Эта петлевая структура сближает вирусную и клеточную мембраны, способствуя их слиянию с последующим проникновением вирусного капсида [29] [30] [31].
[/su_spoiler]
Приблизительно 50% вирусов ВИЧ-1 подтипа B (самая распространенная в США и Европе) высокочувствительны как к теропавимабу, так и зинлирвимабу с 90-процентной ингибирующей концентрацией (IC90) ≤ 2 мкг/мл, и свыше 90% — либо к теропавимабу, либо к зинлирвимабу [32].
Благодаря модификациям Fc-доменов теропавимаба и зинлирвимаба они располагают продленным периодом полувыведения, позволяющим осуществлять дозирование 1 раз в 6 месяцев.
Gilead Sciences создала абсолютную защиту от заражения ВИЧ двумя инъекциями в год. Ленакапавир: самый полный обзор в мире.
КЛИНИЧЕСКИЕ ПОДРОБНОСТИ
Клиническое исследование NCT04811040 фазы Ib (рандомизированное, слепое, многоцентровое) пригласило ВИЧ-инфицированных взрослых пациентов (n=20), находящихся в статусе вирусной супрессии (вирусная нагрузка РНК ВИЧ-1 < 50 копий/мл или неопределяемая) благодаря следованию пероральной АРТ на протяжении не менее чем полутора лет.
Среди основных требований: число T-клеток CD4+ ≥ 500 клеток/мкл; высокая чувствительность ВИЧ-1 одновременно к двум изучаемым нейтрализующим антителам широкого спектра действия: IC90 ≤ 2 мкг/мл.
Участники, прекратившие прием любых АРТ-препаратов, однократно получили подкожно ленакапавир 927 мг (вместе с нагрузочной пероральной 600-мг дозой в 1-й и 2-й дни), внутривенно теропавимаб 30 мг/кг и внутривенно зинлирвимаб 10 мг/кг или 30 мг/кг.
По прошествии 26 недель наблюдений статус вирусной супрессии был зарегистрирован для 90% (n=18/20) испытуемых [1].
Один пациент в группе 10-мг/кг зинлирвимаба столкнулся с вирусной отдачей (уровень РНК ВИЧ-1 ≥ 50 копий/мл), но после перехода на стандартную АРТ вирусная нагрузка была нормализована. Второй пациент в группе 30-мг/кг зинлирвимаба вышел из исследования на 12-й неделе, будучи в статусе вирусной супрессии.
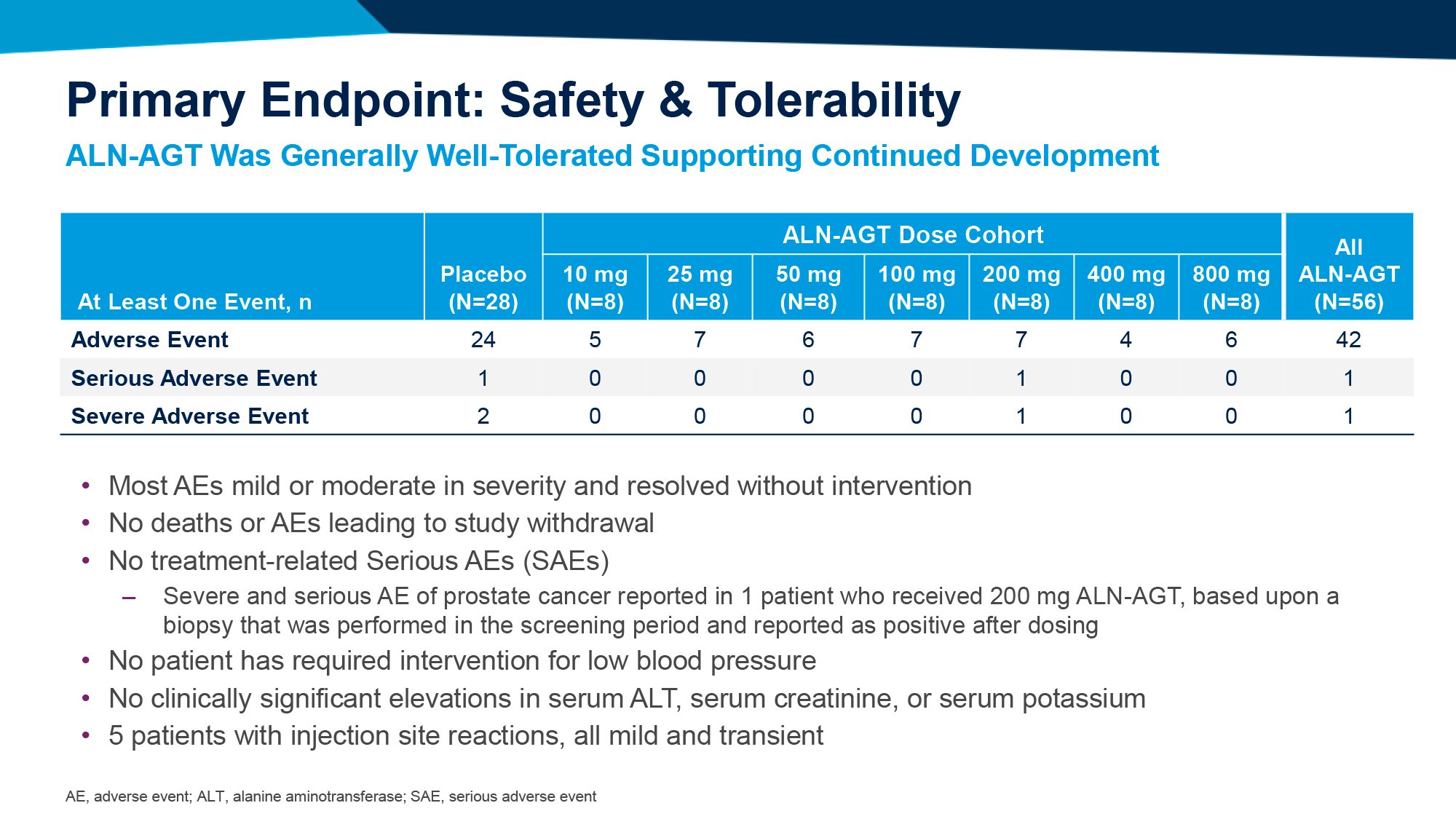
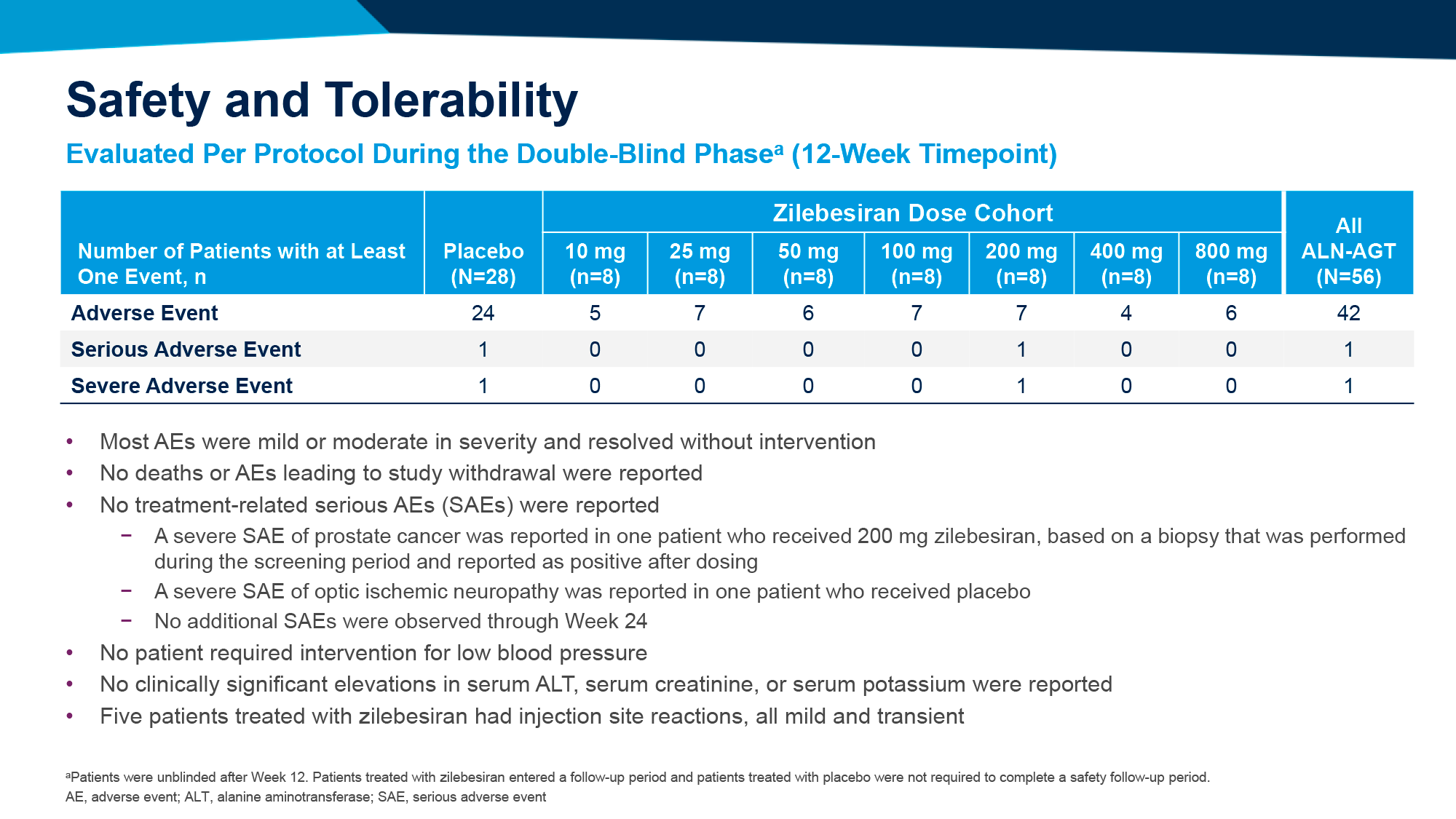
Профиль безопасности экспериментальной терапии благоприятствовал. Серьезных нежелательных явлений (НЯ) отмечено не было. Среди наиболее распространенных НЯ: реакции по месту подкожного введения ленакапавира.
Отдельно исследователи изучили эффективность схемы LEN + TAB + ZAB среди пациентов (n=10) в статусе вирусной супрессии, ВИЧ-1 которых был высокочувствителен только к одному из антител.
После 26 недель наблюдений вирусная супрессия была подтверждена для 80% (n=8/10) участников: 50% (n=2/4) и 100% (n=6/6) в группах 10-мг/кг и 30-мг/кг зинлирвимаба [2].
Если говорить о резистентности среди всех 30 испытуемых, то к антителам ее не было вообще, тогда как к ленакапавиру лекарственная устойчивость развилась лишь у одного человека. Вирусной отдачи в теории можно избежать, если назначать повышенную дозу зинлирвимаба [3].
КОНТРАРГУМЕНТЫ
Несмотря на свою привлекательность, успешное сопряжение нейтрализующих антител широкого спектра действия с низкомолекулярными АРТ-препаратами длительного действия должно преодолеть несколько важных проблем.
В идеале всем лекарственным компонентам такой схемы следует располагать сопоставимыми периодами полувыведения в целях согласованного, одинакового режима дозирования.
Потенциальным ограничением является необходимость вводить антитела путем внутривенной инфузии — разве что если состав и фармакокинетика не позволяют применять их внутримышечно или подкожно. Внутривенный способ назначения по сути отменяет «домашнее», самостоятельное лечение пациентами.
Куда более важно то, что ни одно из существующих антител, в отличие от высокоэффективных АРТ-препаратов, не способно нейтрализовать 100% штаммов ВИЧ [1]. То есть сохраняется риск, что в какой-то момент времени терапия будет осуществляться по факту одним лишь АРТ-препаратом. В свою очередь это несет риск развития резистентности: вирусологическая неудача для всех трех доступных на сегодня АРТ-препаратов длительного действия — CAB, RPV и LEN — была связана как раз с появлением лекарственной устойчивости [2] [3] [4] [5].
Таким образом, для выявления пациентов, которым подойдут подобные новаторские комбинированные схемы лечения, требуется тестирование, подтверждающее чувствительность к нейтрализующим антителам широкого спектра действия. А поскольку в большинстве сценариев люди, живущие с ВИЧ, уже придерживаются супрессивного АРТ-режима, тестирование должно основываться на анализе провирусной ДНК из латентно инфицированных мононуклеарных клеток периферической крови (PBMC). И хотя было обнаружено хорошее соответствие между тестированием вируса в плазме до начала АРТ и в PBMC после начала АРТ [6], корреляция с клиническим ответом на антитела была неоднозначной [7] [8].
Всё вышесказанное не только замедляет набор участников в клинические испытания, но и создает существенные логистические проблемы для широкого внедрения данных терапевтических комбинаций, даже если они окажутся безопасными и эффективными.
Проблема может быть решена разработкой АРТ-препаратов длительного действия с более высоким барьером резистентности или антител, к которым достоверно чувствительны почти все штаммы ВИЧ. Необходимо также продемонстрировать хотя бы нехудшую эффективность по сравнению с существующими АРТ-схемами, плюс показать экономическую обоснованность. Наконец, в целях обеспечения равного доступа в глобальном масштабе требуется оптимизация производственного цикла для снижения себестоимости наряду с подготовкой более совершенных рецептур, пригодных для самостоятельного применения.
ЧТО ДАЛЬШЕ
Осенью 2024 года завершится клиническое исследование NCT05729568 фазы II среди взрослых пациентов (n=83) в статусе вирусной супрессии, которые были переведены с пероральной АРТ-схемы на экспериментальную LEN + TAB + ZAB. ВИЧ-1 участников должен быть чувствительным к обоим bNAb. Будут сняты конечные точки эффективности по прошествии 26 и 52 недель наблюдений.
«Виив хелскеа» (ViiV Healthcare) пытается усовершенствовать доконтактную профилактику (PrEP) инфекции вируса иммунодефицита человека (ВИЧ) так, чтобы она стала максимально удобной для пациента.
ОСНОВНЫЕ ФАКТЫ
Пероральные PrEP-препараты очень эффективно защищают от заражения ВИЧ, но их приходится принимать на постоянной основе. Это ложится ощутимым бременем на приверженность профилактике: легко забыть об очередной дозе лекарства— а затем горько сожалеть.
Не так давно появился инъекционный способ предупреждения ВИЧ-инфекции, предполагающий внутримышечные инъекции 1 раз в 2 месяца.
В обозримом будущем PrEP станет еще удобнее: 1 инъекция раз в 4 месяца или даже в полгода.
С учетом специфики конечных потребителей из групп высокого риска заражения ВИЧ, на которых в основном ориентирована PrEP, подобное очень редкое дозирование более чем востребовано.
Открытым остается вопрос высокой цены: пока стоимость инъекционных PrEP-препаратов не станет сравнимой с таковой у пероральных, вряд ли следует рассчитывать на какое-либо ощутимо резкое снижение числа новых случаев ВИЧ-инфекции.
Gilead Sciences создала абсолютную защиту от заражения ВИЧ двумя инъекциями в год. Ленакапавир: самый полный обзор в мире.
ПРЯМАЯ РЕЧЬ
«ВИЧ-сообщество желает иметь доступ к препаратам более длительного действия, и мы уже заложили основу для следующего поколения таких лекарств. Мы делаем всё возможное, чтобы остановить эпидемию ВИЧ».
Кимберли Смит (Kimberly Smith), руководитель отдела исследований и разработок «Виив хелскеа» (ViiV Healthcare).
«Дальнейшее совершенствование АРТ-препаратов длительного действия — шаг к революции в лечении и профилактике ВИЧ».
Кэлун Хань (Kelong Han), ответственный исследователь из «ГлаксоСмитКляйн» (GlaxoSmithKline).
СУТЬ ВОПРОСА
Долгое время предупредить заражение ВИЧ можно было лишь одним фармакологическим способом: каждый день принимать пероральный препарат «Трувада» (Truvada, эмтрицитабин + тенофовир дизопроксил фумарат; FTC/TDF) — сочетание нуклеозидного ингибитора обратной транскриптазы (NRTI) и нуклеотидного ингибитора обратной транскриптазы (NtRTI) авторства «Гилеад сайенсиз» (Gilead Sciences). Для этих целей он был одобрен в середине июля 2012 года [1].
В начале октября 2019 года «Гилеад» расширила PrEP-возможности, предложив ежедневный пероральный «Дескови» (Descovy, эмтрицитабин + тенофовир алафенамид фумарат; FTC/TAF), который, не уступая «Труваде» в защитной эффективности против ВИЧ-инфицирования, характеризуется более безопасным профилем, поскольку оказывает меньшее негативное влияние на кости и почки.
Женщины могут воспользоваться «Дапиринг» (DapiRing, дапивирин; DPV-VR) — вагинальным кольцом, которое на протяжении месяца высвобождает ненуклеозидный ингибитор обратной транскриптазы (NNRTI), который организует ВИЧ-защиту на локальном уровне, то есть противодействует его передаче исключительно при вагинальном сексе. Гибкое силиконовое кольцо, продвигаемое некоммерческой исследовательской организацией Population Council, доступно главным образом в странах Африки с 2021 года [2] [3] [4].
В конце 2021 года из стен «Виив» вышел «Апретуд» (Apretude, каботегравир; CAB-LA) длительного действия, который, назначаясь внутримышечными инъекциями 1 раз в 2 месяца, гарантирует почти абсолютную защиту от ВИЧ.
Инъекция каботегравира раз в два месяца надежно защитит от ВИЧ-инфекции.
«Виив» на достигнутом не останавливается, продолжая экспериментировать с различными рецептурами каботегравира (cabotegravir), ингибитора переноса цепи интегразой (INSTI, INI), в попытках максимально ослабить бремя инъекций. Не исключено, грядущая PrEP-схема позволит вводить каботегравир 1 раз в квартал, полгода или даже год.
БОЛЬШИЕ ЧИСЛА
Повсеместное распространение и внедрение в клиническую практику высокоэффективной антиретровирусной терапии (АРТ) привело к резкому снижению заболеваемости и смертности от ВИЧ. Однако передача вируса продолжает сохраняться. Об этом свидетельствуют ежегодно накапливаемые данные: если в 2000 году количество новых случаев заражения ВИЧ и летальных исходов от осложнений синдрома приобретенного иммунодефицита (СПИД) составляло 2,8 млн и 1,8 млн, то в 2023-м — 1,3 млн и 630 тыс. А вот число людей, живущих с ВИЧ, выросло с 27,2 млн до 39,9 млн человек [1].
АРТ-препараты требуют пожизненного ежедневного перорального приема, что сопровождается как краткосрочными, так и долгосрочными токсическими эффектами. Не исключаемая вероятность необходимости изменения АРТ-схемы, вызванная появлением резистентности, и строгость в соблюдении режима лечения являются серьезными терапевтическими ограничениями [2] [3] [4] [5].
Вот почему поиск наилучших способов предотвращения распространения инфекции — приоритетное направление исследований в области ВИЧ/СПИДа. Поэтапное развитие доконтактной профилактики при помощи пероральных АРТ-препаратов, которым по силам снижать риск заражения на 99% [6], привело к появлению парентеральной формы каботегравира длительного действия: внутримышечная инъекция 1 раз в 2 месяца — и организована почти абсолютная защита от ВИЧ.
КАК ЭТО РАБОТАЕТ
Каботегравир (cabotegravir [CAB], GSK1265744), будучи ингибитором переноса цепи интегразой (INSTI, INI) и являясь структурным аналогом долутегравира (dolutegravir, DTG), блокирует интегразу ВИЧ путем связывания с ее активным сайтом, тем самым предотвращая перенос и интеграцию вирусного генома в ДНК клеток-хозяина. Поскольку это необходимый шаг для репликации вируса, его дальнейшее распространение становится ограниченным [1] [2].
Если усредненный период полувыведения перорального каботегравира составляет 41 час, то у рецептуры каботегравира с пролонгированным высвобождением — 5,6–11,5 недели.
Ноу-хау каботегравира длительного действия (CAB-LA) реализовано фирменной рецептурой водной наносуспензии, содержащей каботегравир в форме свободной кислоты в виде кристаллических частиц субмикронного размера (около 200 нм) [3] [4] [5].
CAB-LA изготовлен из кристаллической формы свободной кислоты каботегравира, которая обладает низкой растворимостью в воде, длительным периодом полувыведения из организма и высокой противовирусной активностью. Эти свойства оптимальны для системы доставки в виде наносуспензии. Благодаря упаковке действующего вещества в виде твердых кристаллов, как наиболее эффективной формы с точки зрения соотношения веса и объема, удалось обеспечить высокую загрузку препарата, что, в свою очередь, минимизировало объем инъекции для заданной дозы.
CAB-LA разработан как депо-препарат с контролируемым растворением: стадией, лимитирующей (ограничивающей) скорость абсорбции (всасывания) препарата, является растворение его частиц в межклеточной жидкости, окружающей депо. Помимо растворимости влияние на растворение и абсорбцию оказывает размер частиц. Уменьшение размера частиц увеличивает площадь поверхности частиц препарата и, следовательно, скорость их растворения. Наночастицы не только улучшают растворение и абсорбцию, но и повышают удобство введения препарата через шприц.
Соответствующая технология NanoCrystal создана «Элан драг текнолоджис» (Elan Drug Technologies), бизнес-подразделением «Элан корпорейшн» (Elan Corporation) [6]. В сентябре 2011 года «Alkermes» (Alkermes) за 960 млн долларов купила«Элан драг текнолоджис» [7] [8].
На базе технологии NanoCrystal выпущено немало лекарственных препаратов с пролонгированным высвобождением. Так, в июле 2009 года Янссен» (Janssen), входящая в состав «Джонсон энд Джонсон» (Johnson & Johnson), предложила «Инвега Сустенна» / «Ксеплион» (Invega Sustenna /Xeplion, палиперидон), атипичный антипсихотик для лечения шизофрении внутримышечной инъекцией 1 раз в месяц [9]. В мае 2015-го появилась усовершенствованная версия палиперидона (paliperidone) в лице препарата «Инвега Тринза» / «Тревикта» (Invega Trinza / Trevicta, палиперидон), назначаемого 1 раз в 3 месяца [10]. В сентябре 2021 года вышел «Инвега Хафьера» / «Байанли» (Invega Hafyera / Byannli, палиперидон), инъекции которого осуществляются 1 раз в полгода [11].
КЛИНИЧЕСКИЕ ПОДРОБНОСТИ
Продолжающееся клиническое исследование NCT05418868 фазы I (нерандомизированное, открытое, многоцентровое) изучает различные рецептуры каботегравира сверхдлительного действия среди здоровых взрослых добровольцев (n=70).
В одной части испытания участникам назначали каботегравир в новой формуляции (CAB-ULA) подкожно (n=16) или внутримышечно (n=32) в дозах 800 мг, 1200 мг или 1600 мг, а затем сравнивали фармакокинетические показатели с внутримышечной коммерческой рецептурой каботегравира 200 мг/мл (CAB200), уже одобренной под брендом «Апретуд» для PrEP-задач.
Как выяснилось, во-первых, максимальная концентрация CAB-ULA в плазме крови (Cmax), независимо от способа введения, оказалась ниже, чем у CAB200 при той же дозе, что указывает на более медленную абсорбцию (всасывание) CAB-ULA [1].
Во-вторых, прогнозируемый период полувыведения (T1/2), отражающий время, в течение которого препарат находится в организме, при подкожном и внутримышечном введении CAB-ULA получился в 6 и 2 раза продолжительнее, чем период полувыведения CAB200 [2].
Согласно предварительным выводам фармакокинетического моделирования, CAB-ULA, применяемый внутримышечно в дозе 1600 мг/3 мл каждые 4 месяца (или даже реже), потенциально способен обеспечить лекарственную экспозицию на уровне, аналогичном или выше того, который предоставляет «Апретуд», назначаемый в дозе 600 мг/3 мл каждые 2 месяца.
CAB-ULA характеризовался приемлемой переносимостью. Не выявлено нежелательных явлений (НЯ), которые привели бы к прекращению исследования. Наиболее распространенными НЯ были реакции в месте введения препарата, в большинстве случаев носившие легкую степень выраженности и продолжавшиеся не дольше недели.
Рецептуры CAB-ULA с использованием рекомбинантной человеческой гиалуронидазы (rHuPH20) в дозе 10000 МЕ себя не оправдали по двум причинам: T1/2 был сравнимым с таковым у CAB200 (то есть нельзя говорить о возможности более редкого дозирования), НЯ проявлялись в тяжелой форме (эритема с некрозом).
КОНТРАРГУМЕНТЫ
Доконтактная профилактика — чрезвычайно эффективный способ предупредить ВИЧ-инфицирование, но только при условии, что прием PrEP-препарата осуществляется на регулярной основе, то есть на момент потенциального заражения в организме присутствует достаточная концентрация лекарственных веществ. Если человек всё же заражается, это означает, что профилактика проводилась нерегулярно, спорадически, время от времени, с перерывами.
Прорывные инфекции
В очень и очень редких случаях ВИЧ-инфицирование может произойти в процессе следования стабильным курсом PrEP, то есть при достаточном уровне лекарства в организме. В научной литературе зафиксировано совсем немного таких случаев прорывной инфекции — с учетом 6,7 млн людей, когда-либо находившихся на PrEP или продолжающих придерживаться профилактического режима [1].
Так, если говорить об истинной неудаче доконтактной профилактики при помощи «Трувады», в опубликованных историях болезни, обсервационных (наблюдательных) исследованиях и клинических испытаниях было зарегистрировано соответственно 7, 23 и 55 случаев прорывной инфекции. С поправкой на приверженность PrEP искомая частота составила крошечных 0,08% [2].
Считается, что виновником прорыва вируса является половой контакт с человеком со штаммом ВИЧ, который уже выработал устойчивость к лекарственным веществам в составе PrEP и уровень которого достаточно высок для передачи. Как правило, именно мутации резистентности к эмтрицитабину, тенофовиру или обоим были причиной заражения на фоне «Трувады».
Стоит отметить, что в некоторых случаях у людей была венерическая лимфогранулема прямой кишки (разновидность хламидий), которая, есть мнение, приводила к воспалению тканей с последующим образованием локальных ее участков, более уязвимых для ВИЧ.
Случаев прорывной инфекции на фоне «Дескови» зафиксировано не было. Возможно, потому что он относительно недавно внедрился в клиническую практику либо обеспечивает лучшую защиту даже при неоптимальной приверженности.
В ходе клинической проверки «Апретуда» было отмечено 6 случаев истинной неудачи PrEP (частота 0,13%) — все они в связи с резистентностью к каботегравиру [3].
У некоторых людей, столкнувшихся с заражением на фоне «Апретуда», уровень лекарственного вещества внезапно снижался после инъекции. Получается, им требовалось больше времени, чтобы каботегравир длительного действия достиг целевых тканей (например, ректальных и вагинальных), где он формирует защиту. Как бы то ни было, результаты клинических испытаний продемонстрировали, что каботегравир длительного действия значительно эффективнее пероральных PrEP в задаче защиты от ВИЧ-инфицирования, потому что участники существенно реже пропускали очередную дозу.
Настоящей неожиданностью стал первый случай вне клинических испытаний, когда один человек, придерживавшийся адекватного приема «Апретуда», всё же заразился ВИЧ по прошествии 91 дня после перехода с «Трувады». История болезни поразительна глубоким перверсивным поведением и сексуальной распущенностью. Не исключено, уязвимость в период перехода с одного PrEP на другой и выраженная девиантность стали причиной провала профилактики [4] [5] [6].
28-летний индивидуум с биологическим мужским полом, относящий себя к небинарным людям и предпочитающий местоимение «они», вёл сверхактивную половую жизнь с мужчинами цисгендерной идентичности (20–30 новых контактов ежемесячно) и одним основным ВИЧ-положительным партнером (с мутациями резистентности к NRTI и INSTI и неопределяемым уровнем вируса на фоне АРТ), практикуя оральный и анальный секс без презерватива и рецептивный анальный секс с фистингом, а также самоназначая множественные внутримышечные инъекции тестостерона. За 6 месяцев до столкновения с прорывной инфекции был диагностирован вторичный сифилис и оспа обезьян.
Синдром раннего вирусного подавления длительного действия
Имеются некоторые опасения относительно ВИЧ-инфекции, приобретенной во время приема каботегравира длительного действия или до его начала, но не выявленной. Она характеризуется клинической и вирусологической картиной, отличной от острой ВИЧ-инфекции (AHI), которая наблюдается в случае провала пероральной PrEP и которая обычно протекает симптоматично и легко обнаруживается традиционными лабораторными тестами.
Синдром раннего вирусного подавления длительного действия (LEVI) — новый термин, введенный для описания уникальных особенностей ВИЧ-инфекции в контексте каботегравира длительного действия: вялотекущая вирусная репликация, отсроченное выявление с помощью стандартных тестов четвертого поколения, повышенный риск лекарственной устойчивости, минимальные симптомы или их отсутствие [7].
Особую тревогу вызывает резистентность к INSTI, поскольку большинство международных руководств рекомендуют схемы на основе INSTI в качестве первой линии лечения ВИЧ-инфекции [8] [9] [10] [11]. То есть придется прибегать к альтернативной АРТ: например, трехкомпонентному режиму с усиленным дарунавиром — до момента получения результатов тестирования на генотипическую резистентность.
Ввиду вышесказанного следует прибегать к скорректированной стратегии скрининга на ВИЧ: проводить тестирование на РНК ВИЧ в течение недели до начала назначения каботегравира длительного действия, а затем при каждой очередной инъекции, а также ежеквартально на протяжении 12 месяцев после прекращения курса PrEP [12].
Чувствительные РНК-тесты способны выявить большинство новых инфекций до развития основных мутаций резистентности к INSTI [13]. Однако их ограниченная доступность и более высокая стоимость ставит барьеры для широкого доступа к каботегравиру длительного действия, особенно в странах с низким и средним уровнем дохода. Впрочем, даже при отсутствии подобных тестов, основанных на амплификации нуклеиновых кислот (NAAT), повышенный риск резистентности к INSTI компенсируется значительным снижением числа новых ВИЧ-инфекций [14] [15].
Длительный скрининг после прекращения PrEP-инъекций необходим из-за риска развития резистентной ВИЧ-инфекции в период так называемого фармакокинетического хвоста, когда остаточная концентрация каботегравира в плазме, пусть даже сниженная до недостаточного для защиты уровня, всё еще может оказать селективное давление. В этот период рекомендовано назначать пероральные PrEP-препараты [16].
Нежелательные явления
Наблюдаемый рост новых случаев гипертонии при приеме «Апретуда» требует дополнительного изучения и особой бдительности по мере расширения его PrEP-охвата. В литературе всё чаще высказываются предположения о связи INSTI с повышением артериального давления, инсулинорезистентностью, нарушением метаболизма, сердечно-сосудистым риском [17].
Цена
Немаловажен вопрос цены каботегравира длительного действия.
В США одна инъекция «Апретуда» обходится в 3927 долларов (это полная стоимость, не учитывающая скидки и дисконты и взимаемая в случае отсутствия покрытия медицинской страховкой) [18].
Согласно экспертным оценкам, без оглядки на всю перспективность «Апретуда», чтобы его внедрение оказалось финансово оправданным в странах с низким и средним уровнем дохода и высокой распространенностью ВИЧ, цена должна быть разумной и не превышать двойную стоимость двухмесячного запаса «Трувады»: к примеру, укладываться в пределы 9–15 долларов за инъекцию [19].
Согласно оценочному анализу, генерические компании могут производить каботегравир длительного действия по цене 30–40 долларов за годовой профилактический курс. При условии умеренного спроса (800 тыс. потребителей ежегодно) цена снизится до 14–16 долларов. Эти суммы не учитывают капитальные затраты и расходы на разработку в размере 8–10 млн долларов; в смету входит стоимость оборудования, испытаний биоэквивалентности, создания конечной рецептуры [20].
В июле 2022 года «Виив» лицензировала каботегравир длительного действия Патентному фонду лекарственных средств ЮНИТЭЙД (MPP), целью которого является расширение доступа бедных стран к лекарственным препаратам. Девяти десяткам государств (из стран бывшего СССР это Киргизия, Таджикистан, Узбекистан, Украина) дозволено реализовывать на своей территории дженерик «Апретуда» для PrEP. Копии начнут появляться не ранее 2027 года: генерическим фармкомпаниям, включая «Ауробиндо фарма» (Aurobindo Pharma), «Ципла» (Cipla) и «Майлан» (Mylan), которым переданы соответствующие технологии производства, должны не только наладить выпуск, но и подготовить регистрационные досье для местных регуляторов и Всемирной организации здравоохранения (ВОЗ) [21].
Каботегравир длительного действия зарегистрирован в России под брендом «Вокабриа» (Vocabria) в конце 2022 года, но разрешен только для лечения ВИЧ-инфекции [22]. По состоянию на начало августа 2024 года в свободной продаже этого препарата нет.
ЧТО ДАЛЬШЕ
Параллельно «Виив» раскрыла предварительные подробности о новом пролекарстве каботегравира, которое в теории позволит осуществлять еще более редкое дозирование в целях профилактической защиты от ВИЧ-инфицирования: 1 или 2 инъекции в год [1].
Каботегравира стеарат (cabotegravir stearate, M2CAB) формирует локальные и распределенные по макрофагам депо действующего вещества, что приводит к фармакокинетике типа «флип-флоп» — нетипичной ситуации, когда скорость абсорбции препарата (или скорость его поступления в кровь) медленнее скорости его элиминации. M2CAB изучается в рецептуре инъекционной суспензии для внутримышечного введения (XVIR-110) [2] [3].
Согласно доклинической проверке на животных (крысах и собаках), однократное назначение XVIR-110 обеспечило низкую экспозицию пролекарственного M2CAB, но высокую и персистирующую экспозицию лекарственного каботегравира в течение всего периода исследования (7 и 5 месяцев соответственно). Предполагается, что концентрации, превышающие 90-процентную ингибирующую концентрацию с коррекцией на связывание с белками (PB-IC90), сохранятся на протяжении более чем 12 месяцев. Если перенести собранные модельные данные на человеческий организм, получается, что XVIR-110 можно будет назначать 1 раз в 6 или даже 12 месяцев.
XVIR-110 характеризовался приемлемой переносимостью, без признаков дозолимитирующих реакций по месту введения. Экспериментальный препарат продемонстрировал менее выраженные ранние микроскопические изменения после инъекции, включая некроз тканей, воспаление и инфильтрацию иммунными клетками, если сравнивать с коммерческим каботегравиром в эквивалентных дозах.
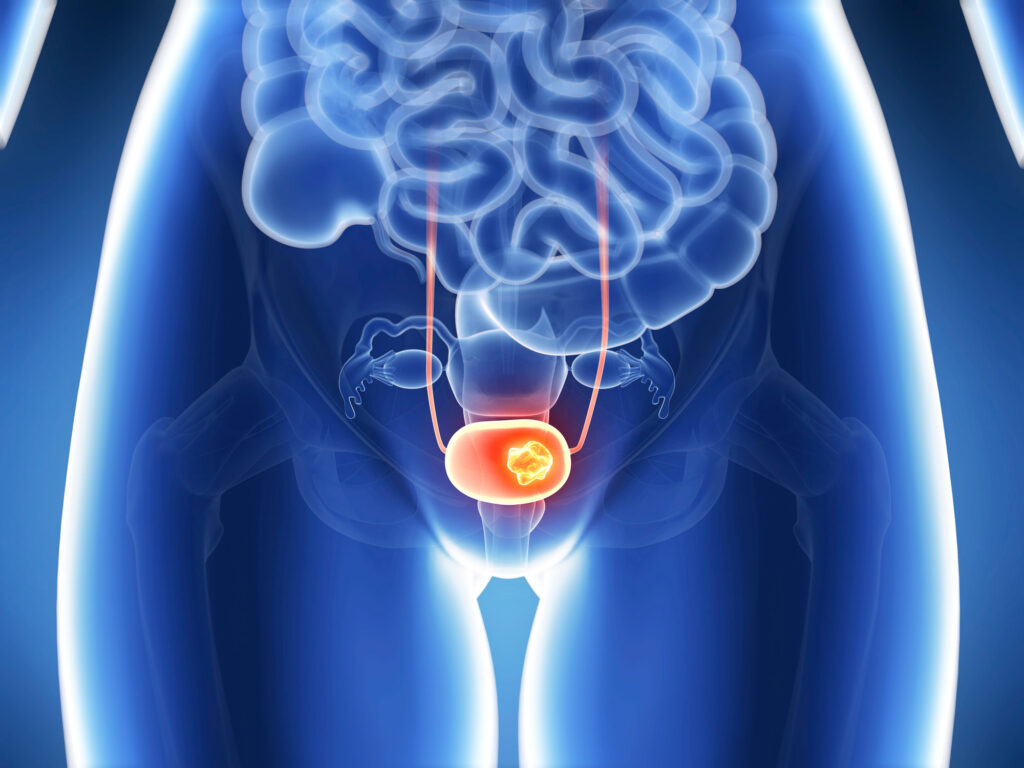
→ Немышечно-инвазивный рак мочевого пузыря (НМИРМП) встречается в три четверти случаев онкологического заболевания этого органа. → Первоочередное лечение НМИРМП предполагает трансуретральную резекцию (ТУР) всей видимой опухоли. → В целях профилактики рецидива назначают продолжительную иммунотерапию бациллой Кальметта — Герена (БЦЖ), которая весьма эффективна. → Однако в ряде случаев НМИРМП перестает реагировать на БЦЖ-вакцину, и тогда варианты дальнейшего консервативного лечения становятся резко ограниченными. → Фармотрасль усиленно работает над новыми способами лечения НМИРМП после провала БЦЖ-иммунотерапии.
СУТЬ ВОПРОСА
Во всём мире рак мочевого пузыря занимает десятое место среди наиболее часто диагностируемых видов онкологических заболеваний [1] [2]. Это онкологическое заболевание главным образом пожилых людей: средний возраст постановки диагноза составляет 69–71 лет [3].
В развитых странах рак мочевого пузыря преимущественно (в 90% случаев) характеризуется уротелиальной гистологией (ранее относился к переходно-клеточному раку) [4].
Уротелиальная карцинома мочевого пузыря в 70% случаев классифицируется как немышечно-инвазивный рак мочевого пузыря (НМИРМП), то есть как рак мочевого пузыря без прорастания в мышечный слой [5] [6].
После трансуретральной резекции (ТУР) всей видимой опухоли, как первоочередного хирургического лечения НМИРМП, 40–80% пациентов сталкиваются с рецидивом в течение последующих 6–12 месяцев, а 10–25% — прогрессируют до мышечно-инвазивного, регионарного или метастатического заболевания [7].
Таким образом, существует необходимость в способах лечения, дополняющих ТУР и преследующих цель сдерживания заболевания.
СЕГОДНЯ
Немышечно-инвазивный рак мочевого пузыря (НМИРМП), согласно гистологическому стадированию в зависимости от характера роста и глубины инвазии, может быть неинвазивной папиллярной карциномой (Ta), карциномой in situ (CIS; Tis) и с распространением на субэпителиальную соединительную ткань (T1) — соответственно в 70%, 10% и 20% случаев [1] [2].
После трансуретральной резекции (ТУР) всей видимой опухоли НМИРМП применяют, для предупреждения его рецидива или прогрессирования, интравезикальную (внутрипузырную) терапию, которая обеспечивает высокую локальную концентрацию терапевтического препарата внутри мочевого пузыря, потенциально уничтожая оставшиеся жизнеспособные опухолевые клетки.
Интравезикальное введение бациллы Кальметта — Герена (БЦЖ) — живой аттенуированной формы Mycobacterium bovis, возбудителя туберкулеза у крупного рогатого скота, предложенное еще в 1972 году [3] [4], является стандартной адъювантной процедурой, дополняющей ТУР при НМИРМП [5] [6] [7].
В качестве альтернативы БЦЖ-вакцине широко используется ряд химиотерапевтических препаратов, в частности, митомицин (mitomycin), эпирубицин (epirubicin) и гемцитабин (gemcitabine).
Высокую эффективность БЦЖ-иммунотерапии не удалось превзойти никакому из изученных внутрипузырных препаратов [8] [9] [10] [11] [12] [13].
Противоопухолевое действие БЦЖ-вакцины многогранно и включает следующие предполагаемые механизмы: индукция инфильтрата мононуклеарных клеток, состоящего преимущественно из T-клеток CD4+ и макрофагов; повышение экспрессии интерферона гамма (IFNγ); усиление экспрессии интерлейкинов 1, 2, 6, 8 и 12 (IL-1, IL-2, IL-6, IL-8, IL-12), фактора некроза опухоли (TNF) и TNF-связанного индуцирующего апоптоз лиганда (TRAIL); прямое подавление роста опухоли [31] [32] [33] [34] [35] [36].
Внутрипузырные инстилляции БЦЖ-вакцины показаны при высокорисковом немышечно-инвазивном раке мочевого пузыря, под определение которого подпадают CIS (Tis) и высокозлокачественные опухоли Ta или T1. Она также является вариантом для определенных пациентов с промежуточным риском заболевания [14] [15] [16] [17]. Согласно метаанализам, поддерживающее лечение при помощи БЦЖ на протяжении не менее чем одного года снижает риск рецидива или прогрессирования по сравнению с химиотерапией [18] [19] [20] [21].
БЦЖ-иммунотерапия способствует отсрочиванию прогрессирования опухоли до более распространенной стадии, снижению риска необходимости в последующей радикальной цистэктомиии (хирургическом удалении мочевого пузыря) и продлению общей выживаемости [19] [20] [21] [22] [23] [24] [25].
Однако в 40–50% случаев регистрируется провал БЦЖ-иммунотерапии, то есть, если упрощенно, обнаружение высокозлокачественной опухоли в ходе лечения или после него [18] [26] [27] [28] [29].
Дальнейшее лечение НМИРМП обычно предполагает радикальную цистэктомию, которая, сопровождающаяся заболеваемостью и смертностью, резко ухудшает качество жизни [30].
Вот почему необходимы варианты лечения немышечно-инвазивного рака мочевого пузыря, которые могли бы применяться после неудачи БЦЖ-иммунотерапии и которые приводили бы к устранению или отсрочиванию необходимости в радикальной цистэктомии.
НОВИНКИ
В начале января 2020 года «Китруда» (Keytruda, пембролизумаб), блокатор PD-1 авторства «Мерк и Ко» (Merck & Co.),получил разрешение Управления по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) для лечения высокорискового немышечно-инвазивного рака мочевого пузыря (НМИРМП) с карциномой in situ (CIS) с папиллярными новообразованиями (или без них), не реагирующего на назначение бациллы Кальметта — Герена (БЦЖ), — у пациентов, не подходящих для прохождения цистэктомии или отказавшихся от нее.
Пембролизумаб позволит избежать радикальной цистэктомии.
В середине декабря 2022 года FDA одобрило«Адстиладрин» (Adstiladrin, надофараген фираденовек) — генно-терапевтическое лечение, разработанное швейцарской «Ферринг фармасьютикалс» (Ferring Pharmaceuticals) для применения при таком же показании НМИРМП.
Надофараген фираденовек (nadofaragene firadenovec, rAd–IFN/Syn3) построен на основе нереплицирующегося рекомбинантного аденовирусного вектора (rAd), кодирующего ген интерферона альфа-2b (IFNα-2b) человека. Последний, выступая естественным регулятором активности иммунной системы, активирует транскрипцию и трансляцию генов, ответственных за продукцию определенных ферментов, подавление клеточной пролиферации, а также иммуномодулирующую активность, включая фагоцитарную активность макрофагов и усиление специфической цитотоксичности лимфоцитов в отношении клеток-мишеней [1] [2] [3] [4] [5].
Надофараген фираденовек позволит избежать радикальной цистэктомии.
В конце апреля 2024 года на сцену вышел «Анктива» (Anktiva, ногапендекин альфа инбакицепт), предложенный «Имьюнитибайо» (ImmunityBio) для лечения такой же категории пациентов с НМИРМП. «Анктива», в отличие от монотерапии «Китрудой» или «Адстиладрином», назначается совместно с БЦЖ.
Ногапендекин альфа инбакицепт (nogapendekin alfa inbakicept, N-803, ALT-803) представляет собой антительный слитый (гибридный) белок, составленный из мутированного (N72D) человеческого интерлейкина 15 (IL-15), Sushi-домена внеклеточной области альфа-субъединицы рецептора IL-15 (IL-15Rα), Fc-области иммуноглобулина G1 (IgG1). Получившийся гетеродимерный комплекс является суперагонистом IL-15, который полностью имитирует противоопухолевую эндогенную биологию интерлейкина 15, добавляя к нему стабильность и продленный период полувыведения. Связывание ногапендекина альфа инбакицепта с рецепторами IL-15, экспрессирующими на T-клетках CD4+ and CD8+ и естественных киллерных (NK) клетках, приводит к активации и экспансии T-клеточного иммунитета, причем без стимуляции иммуносупрессивных регуляторных T-клеток (Treg), сдерживающих иммунный ответ [6] [7] [8].
ЗАВТРА
Фармотрасль продолжает разрабатывать новые способы лечения немышечно-инвазивного рака мочевого пузыря (НМИРМП) высокого риска, на который не подействовала иммунотерапия бациллой Кальметта — Герена (БЦЖ). Цель прозрачна: предложить консервативное лечение, которое максимально длительно откладывает необходимость в радикальной цистэктомии.
В своем подавляющем большинстве экспериментальные препараты применяются интравезикально (внутрипузырно).
ХИМИОТЕРАПИЯ
В продолжающемся клиническом испытании SunRISe-1 (NCT04640623) фазы IIb, организованном «Янссен» (Janssen) в составе «Джонсон энд Джонсон» (Johnson & Johnson), экспериментальная система TAR-200 (в виде 5-см кренделька) для таргетного и продолжительного (приблизительно 3 недели) локального высвобождения химиотерапевтического гемцитабина (gemcitabine) в мочевом пузыре обеспечила полный ответ у 83% (71–91) пациентов. Вероятность длительности ответа (DOR) на протяжении 1 года составила 75% (50–88) [1] [2].
Перспективной выглядит химиотерапевтическая тройка из кабазитаксела (cabazitaxel), гемцитабина (gemcitabine) и цисплатина (cisplatin), внутрипузырное последовательное введение которых, проверенное в клиническом исследовании фазы I, установило частоту безрецидивной выживаемости (RFS) на протяжении 12 и 24 месяцев на уровне 83% и 64%, притом что эти показатели оказались еще лучше, 100% и 83%, при получении максимальных доз [3]. К концу 2024 года ожидается завершение клинического испытания NCT02202772 фазы II [4].
Не следует также забывать об альтернативе БЦЖ-вакцине, которая давно находится в дефиците, в лице химиотерапевтических гемцитабина (gemcitabine) и доцетаксела (docetaxel), применяемых интравезикально и последовательно. Так, частоты 12- и 24-месячной RFS при назначении БЦЖ составили 71% (64–78) и 69% (62–76), а при химиотерапии — 85% (78–91) и 81% (72–87) [5] [6]. Продолжается клиническое исследование BRIDGE (NCT05538663) фазы III, которое призвано раскрыть нюансы выбора между БЦЖ-иммунотерапией и химиотерапией в ходе первоочередного лечения немышечно-инвазивного рака мочевого пузыря [7].
ТАРГЕТНАЯ ТЕРАПИЯ
«Янссен» (Janssen) в составе «Джонсон энд Джонсон» (Johnson & Johnson) подтвердила терапевтическую состоятельность эрдафитиниба (erdafitinib), тирозинкиназного ингибитора рецепторов 1–4 фактора роста фибробластов (FGFR1–4), альтерации которых, будучи онкодрайверными, обнаруживаются в 50–80% случаев НМИРМП [1] [2].
Во-первых, клиническое исследование THOR-2 (NCT04172675) фазы II, сравнившее пероральный эрдафитиниб с интравезикальной химиотерапией, показало его превосходство в том, что касается продления выживаемости без прогрессирования (PFS) [3].
Во-вторых, TAR-210, экспериментальное устройство для внутрипузырного локального высвобождения эрдафитиниба на протяжении 3 месяцев, справилось с клиническим исследованием NCT05316155 фазы I, продемонстрировав, согласно промежуточным результатам, способность сдерживать рецидив [4] [5].
Сейчас пероральный «Балверса» (Balversa, эрдафитиниб)разрешен для лечения местнораспространенного или метастатического рака мочевого пузыря с чувствительными к нему FGFR3-альтерациями, прогрессировавшего после или во время хотя бы одной линии системной терапии. В планах «Джонсон энд Джонсон» стоит расширение спектра показаний «Балверсы».
Эрдафитиниб для лечения местнораспространенной или метастатической уротелиальной карциномы с FGFR3-альтерациями.
Промежуточные результаты клинического исследования EV-104 (NCT05014139) фазы I указали на потенциальную возможность интравезикального применения энфортумаба ведотина (enfortumab vedotin) в лечении НМИРМП после провала БЦЖ-иммунотерапии [6].
Этот конъюгат моноклонального антитела против нектина-4, известный как «Падцев» (Padcev) и несущий цитотоксический монометилауристатин E (MMAE), разработанный «Астеллас фарма» (Astellas Pharma) и «Сиджин» (Seagen), которую купила «Пфайзер» (Pfizer), сейчас назначается внутривенно в первоочередном лечении местнораспространенного или метастатического уротелиального рака.
Комбинация из энфортумаба ведотина и пембролизумаба продлит жизнь при неоперабельной уротелиальной карциноме.
Энфортумаб ведотин избирательно связывается с нектином-4 — иммуноглобулиноподобной молекулой клеточной адгезии и опухолеассоциированным антигеном, также известным как белок 4, связанный с рецептором полиовируса (PVRL4, PRR4), который сверхэкспрессирует на поверхности злокачественных клеток при различных солидных опухолях. После интернализации энфортумаба ведотина и протеолитического расщепления линкера происходит связывание MMAE с тубулином, что ингибирует полимеризацию последнего. Это приводит к остановке фазы G2/M клеточного цикла и индуцированию апоптоза опухолевых клеток, сверхэкспрессирующих нектин-4 [7] [8] [9] [10] [11].
Китайская «Римиджин» (RemeGen) сделала ставку на диситамаб ведотин (disitamab vedotin, RC48) — конъюгат моноклонального антитела, таргетированный на рецептор 2 эпидермального фактора роста 2 (HER2) и несущий цитотоксический монометилауристатин E (MMAE). Оценочная частота опухолевой HER2-экспрессии (совокупно IHC 2+ и 3+) при немышечно-инвазивном раке мочевого пузыря может доходить до 50% [12].
Сейчас этот препарат, одобренный под брендовым названием «Айдикси» (Aidixi), применяется внутривенно в лечении HER2-положительного местнораспространенного или метастатического рака желудка и уротелиального рака, ранее прошедших терапию. В августе 2021 года «Сиджин» приобрела у «Римиджин» мировые права (за исключением Азиатско-Тихоокеанского региона) на диситамаб ведотин [13].
Согласно ретроспективным клиническим данным, среди пациентов, получивших интравезикальные инстилляции диситамаба ведотина, частота 12-месячной RFS составила 100%, тогда как среди больных, которые прошли БЦЖ-иммунотерапию, этот показатель получился равным 58%. Впрочем, статистически значимого расхождения не зафиксировано (p=0,22) [14].
Запущены клинические исследования NCT06378242 фазы I/II, NCT05957757 фазы II и NCT05943379 фазы II, которые проверяют соответственно мононазначение диситамаба ведотина, его комбинацию с PD-1-блокатором «Тевимбра» (Tevimbra, тислелизумаб) авторства китайской «Бейджин» (BeiGene) и его сочетание с гемцитабином (gemcitabine). Первое испытание организовано среди пациентов, прежде не получавших БЦЖ-вакцину, второе и третье — уже прошедших ее курс.
Экспериментальная фотодинамическая терапия «Рутеррин» (Rutherrin), обкатываемая канадской «Тералейз текнолоджис» (Theralase Technologies) в ходе клинического исследования NCT03945162 фазы II, продемонстрировала, согласно промежуточным данным, вывод 63% (n=38/59) пациентов к статусу CR [15].
После интравезикального введения «Рутеррина», который сочетает производное рутения TLD-1433 с гликопротеином трансферрином, осуществляется его активация лазерным излучением. «Рутеррин», преимущественно поглощенный раковыми клетками (они характеризуются повышенной экспрессией рецепторов трансферрина), начинает вырабатывать синглетный (атомарный) кислород и кислородные радикалы — активные формы кислорода, вызывающие в этих клетках окислительный стресс с последующей их гибелью [16] [17] [18] [19] [20] [21].
ИММУНОТЕРАПИЯ
Продолжается клиническое исследование HOPE-04 (ChiCTR2200059970) фазы II, проверяющее гипотезу оправданности короткого курса облучения в сочетании с внутривенным торипалимабом (toripalimab), блокатором PD-1 китайской «Шанхай Цзюньши байосайенсиз» (Shanghai Junshi Biosciences), одобренным под брендом «Локторзи» (Loqtorzi) для лечения рака носоглотки [1].
Китайская «Цзянсу Симсиа фармасьютикал» (Jiangsu Simcere Pharmaceutical) придумала добавлять к БЦЖ-вакцине (или использовать монотерапевтически) экспериментальный противоопухолевый и иммуностимулирующий слитый белок SIM0237, составленный из моноклонального антитела против PD-L1 и комплекса со сниженной потентностью, включающего интерлейкин 15 (IL-15) и Sushi-домен внеклеточной области альфа-субъединицы рецептора IL-15 (IL-15Rα) [2] [3]. Клиническое исследование NCT06186414 фазы II продолжается.
БАКТЕРИОТЕРАПИЯ
Экспериментальный TARA-002, изучаемый «Протара терапьютикс» (Protara Therapeutics), продемонстрировал обнадеживающие 3-месячные результаты клинических исследований ADVANCED-1 (NCT05085977) фазы Ia/Ib и ADVANCED-2 (NCT05951179) фазы II: совокупный выход к CR-статусу составил 38% (n=6/16), варьируя в широких пределах в зависимости от особенностей заболевания и его предшествовавшего лечения [1].
TARA-002 — иммунопотенцирующий препарат широкого спектра действия, по своему механизму действия схожий с БЦЖ-вакциной, активирующий врожденные и адаптивные иммунные клетки, а также непосредственно уничтожающий опухолевые клетки. TARA-002 разработан на основе того же клеточного банка из генетически разнообразных пиогенных стрептококков (Streptococcus pyogenes) группы A, что и «Пицибанил» (Picibanil, OK-432), продвигаемый японской «Шугай фармасьютикал» (Chugai Pharmaceutical) и успешно применяющийся в Японии и на Тайване с 1975 года для лечения лимфангиомы (лимфатической мальформации) [2] [3] [4] [5].
В начале февраля 2024 года началось клиническое исследование PARADIGM-1 (NCT06181266) фазы I/Ib, тестирующее экспериментальный ZH9, который британская «Прокариум» (Prokarium) создала на основе генетически модифицированного штамма ZH9 аттенуированной бактерии Salmonella enterica подвида enterica серовара Typhi, вызывающей брюшной тиф [6] [7].
По сути «Прокариум», ратующая за идею альтернативы БЦЖ-вакцине, лицензировала наработки Университетской больницы Лозанны (CHUV) [8], специалисты которого неоднократно демонстрировали терапевтическую оправданность данного подхода на примере интравезикальной рецептуры пероральной тифозной вакцины «Вивотиф» (Vivotif, Ty21a) [9] [10] [11] [12] [13].
ВИРУСНАЯ ТЕРАПИЯ
«Си-джи онколоджи» (CG Oncology) предложила атаковать НМИРМП онколитическим вирусом кретостимоген гренаденорепвек (cretostimogene grenadenorepvec, CG0070), который избирательно реплицируется в опухолевых клетках с дефектом сигнального пути белка ретинобластомы (RB). При раке мочевого пузыря его опухолевые клетки почти всегда несут RB-мутации. Кретостимоген гренаденорепвек работает путем прямого лизиса опухоли и через механизмы иммуногенной гибели раковых клеток [1] [2] [3] [4] [5].
Согласно промежуточным данным клинических испытаний, кретостимоген гренаденорепвек предоставил лучшие исходы, если говорить о частоте полного ответа (CR) — в сравнении с уже одобренными препаратами против НМИРМП, такими как «Китруда» (Keytruda, пембролизумаб), «Адстиладрин» (Adstiladrin, надофараген фираденовек) и «Анктива» (Anktiva, ногапендекин альфа инбакицепт).
Кретостимоген гренаденорепвек: онколитический вирус против уротелиальной карциномы, не реагирующей на иммунотерапевтическую вакцину БЦЖ.
ГЕННАЯ ТЕРАПИЯ
Экспериментальная невирусная иммуноонкологическая генная терапия деталимоген вораплазмид (detalimogene voraplasmid, EG-70) выдала CR на уровне 73% (n=16/22) в продолжающемся клиническом исследовании LEGEND (NCT04752722) фазы I/II [1] [2].
Деталимоген вораплазмид, разработкой которого занимается американо-канадская «Энджин» (enGene), доставляет в эпителий слизистых тканей мочевого пузыря ДНК-плазмиду для локальной экспрессии рекомбинантного одноцепочечного интерлейкина 12 (IL-12) [двух его субъединиц, p40 и p35 ] и двух двухцепочечных РНК-активаторов (VA1 и eRNA11a) сигнального пути индуцируемого ретиноевой кислотой гена 1 (RIG-I), являющегося внутриклеточным регулятором врожденного иммунитета.
Механизм действия деталимогена вораплазмида обращается к индуцированию выработки интерферона гамма (IFNγ), который является сильным противоопухолевым и антиангиогеннным цитокином, и стимулированию мощного воспалительного ответа, который приводит к прямому уничтожению опухолевых клеток, опосредованной цитокинами активации клеток врожденного иммунитета, рекрутингу и перекрестному примированию T-клеток.
РНК-ТЕРАПИЯ
В конце апреля 2024 года китайская «Рактиджен терапьютикс» (Ractigen Therapeutics) получила разрешение Управления по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) на проведение первого клинического испытания экспериментального RAG-01, который построен на базе коротких активирующих РНК (каРНК) и который восстанавливает экспрессию опухолевого супрессора p21, обычно заглушенного в раковых клетках мочевого пузыря [1].
ПРОВАЛЫ
Определенных успехов в лечении высокорискового немышечно-инвазивного рака мочевого пузыря (НМИРМП), который перестал реагировать на иммунотерапию бациллой Кальметта — Герена (БЦЖ), добилась «Сесен байо» (Sesen Bio), ранее называвшаяся «Илевен байотерапьютикс» (Eleven Biotherapeutics) и в марте 2023 года слившаяся с «Карисма терапьютикс» (Carisma Therapeutics) [1].
Экспериментальный «Вицинеум» (Vicineum, опортузумаб монатокс) по прошествии 3 месяцев клинического исследования VISTA (NCT02449239) фазы III вывел 40% (30–51) пациентов с к полному ответу (CR), хотя, когда миновали 12 месяцев, ремиссия сохранилась лишь у 17% (10–26) испытуемых [2].
Опортузумаб монатокс (oportuzumab monatox, VB4-845) — иммунотоксин, состоящий из гуманизированного одноцепочечного вариабельного фрагмента моноклонального антитела против молекулы клеточной адгезии эпителия (EpCAM), конъюгированного с усеченной формой экзотоксина A синегнойной палочки (Pseudomonas aeruginosa). Поскольку при раке мочевого пузыря наблюдается сверхэкспрессия мембранного белка EpCAM, опортузумаб монатокс осуществляет таргетную доставку в опухолевые клектки цитотоксической нагрузки, которая их уничтожает, блокируя белковый синтез [3] [4] [5] [6].
В июле 2022 года «Сесен» свернула программу разработки «Вицинеума» для лечения НМИРМП [7].
Не оправдала себя затея объединения БЦЖ-вакцины с противораковой вакциной «Панвак» (Panvac): исходы клинического исследования NCT02015104 фазы II статистически значимо не отличались от применения только БЦЖ-иммунотерапии [8].
Онковакцина «Панвак» (CEA-MUC-1-TRICOM, CV301, inalimarev–falimarev), разработанная «Терион байолоджикс» (Therion Biologics), состоит из двух векторов: модифицированного осповакцинного вируса и рекомбинантного вируса оспы кур, — они применяются в режиме прайм-буст. Оба вектора кодируют трансгены двух опухолеассоциированных антигенов, муцина 1 (MUC1) и карциноэмбрионального антигена (CEA), и трех костимулирующих T-клетки молекул, B7.1 (CD80), ICAM-1 (CD54) и LFA-3 (CD58). За счет того, что антигенпрезентирующим клеткам (APC) демонстрируются MUC1 и CEA, которые сверхэкспрессированы при карциномах, активируется ответ цитотоксических T-лимфоцитов (CTL). В качестве опционального адъюванта используется гранулоцитарно-макрофагальный колониестимулирующий фактор (GM-CSF) [9] [10] [11] [12] [13] [14].
Не повезло израильской «Анкиано терапьютикс» (Anchiano Therapeutics), в марте 2021 года объединившейся с местной «Кемоумаб» (Chemomab) и теперь называющейся «Кемоумаб терапьютикс» (Chemomab therapeutics): в ноябре 2019 года пришлось остановить программу весьма интересной генной терапии, но не обеспечившей должной частоты ремиссии. В клиническом исследовании CODEX (NCT03719300) фазы II, согласно промежуточному анализу, к CR-статусу вышли 19% (n=3/16) пациентов [15] [16].
Инодифтаген викстеплазмид (inodiftagene vixteplasmid, BC-819) — рекомбинантная ДНК-плазмида, несущая ген цепи дифтерийного токсина A (dT-A) и протомор H19. За счет того, что транскрипционные факторы H19 в избытке представлены в опухолевых клетках, осуществляется активация экспрессии dT-A, который подавляет синтез белков и вызывает гибель опухолевых клеток. Инодифтаген викстеплазмид не содержит гена цепи дифтерийного токсина B (dT-B), что предотвращает передачу цепи dT-A между клетками. Онкофетальный ген H19 высоко экспрессируется эмбриональными и некоторыми злокачественными тканями, но слабо представлен в нормальных взрослых тканях [17] [18] [19] [20] [21].
Клиническая проверка показала, что сочетание препаратов «Опдиво» (Opdivo, ниволумаб) и «Ервой» (Yervoy, ипилимумаб), назначаемое в ходе первоочередного лечения неоперабельного рака печени (гепатоцеллюлярной карциномы), превосходит эффективность, обеспечиваемую терапией в лице препарата «Ленвима» (Lenvima, ленватиниб) или «Нексавар» (Nexavar, сорафениб).
ОСНОВНЫЕ ФАКТЫ
«Бристол-Майерс Сквибб» (Bristol-Myers Squibb) продемонстрировала преимущество сочетания из ниволумаба (nivolumab) и ипилимумаба (ipilimumab), блокаторов PD-1 и CTLA-4, над ленватинибом (lenvatinib) или сорафенибом (sorafenib), тирозинкиназными ингибиторами, продвигаемыми соответственно «Эйсай» (Eisai) / «Мерк и Ко» (Merck & Co.) и «Байер» (Bayer), в ходе перволинейной терапии рака печени.
По отношению к препаратам сравнения иммунноонкологический коктейль снизил риск смерти на 21% и снизил риск прогрессирования заболевания или смерти на 13%.
Этого недостаточно, чтобы опередить нынешний стандарт в лице комбинации из «Тецентрика» (Tecentriq, атезолизумаб) и «Авастина» (Avastin, бевацизумаб) — блокатора PD-L1 и ингибитора VEGF авторства «Рош» (Roche).
Однако в абсолютном исчислении продление общей выживаемости оказалось превосходным.
Регистрационное досье отправлено в адрес регуляторов.
ПРЯМАЯ РЕЧЬ
«Медиана общей выживаемости получилась одной из самых длинных, которые мы когда-либо наблюдали в ходе лечения распространенной гепатоцеллюлярной карциномы. Мы уверены, что разработали новый стандарт лечения».
Питер Галле (Peter Galle), клинический гепатолог из Медицинского центра при Майнцском университете (земля Рейнланд-Пфальц, Германия).
«Отмеченная нами частота уменьшения опухоли — одна из самых высоких среди других вариантов лечения рака печени. Высокий уровень ответа на терапию повышает шансы трансформации заболевания из неоперабельного в поддающееся резекции».
Лаура Гофф (Laura Goff), исполнительный медицинский директор Центра ухода за онкологическими пациентами при Онкологическом центре Вандербильта — Инграма (VICC, шт. Теннесси, США).
«Несмотря на продолжающееся развитие фармакологической науки, прогноз для пациентов с гепатоцеллюлярной карциномой по-прежнему остается плохим. Вот почему важно предложить им новые способы лечения, которые, возможно, помогут».
Дана Уолкер (Dana Walker), вице-президент и руководитель глобальной программы по раку желудочно-кишечного тракта и мочеполовой системы «Бристол-Майерс Сквибб» (Bristol-Myers Squibb).
КЛИНИЧЕСКИЕ ПОДРОБНОСТИ
Клиническое исследование CheckMate 9DW (NCT04039607) фазы III (рандомизированное, открытое, с активным контролем, многоцентровое, международное) пригласило взрослых пациентов (n=668) с распространенной гепатоцеллюлярной карциномой, ранее не проходившей системной терапии.
Участникам назначали либо комбинацию из ниволумаба и ипилимумаба, либо ленватиниб или сорафениб (на выбор исследователя) — до момента прогрессирования заболевания или неприемлемой токсичности.
После наблюдений на протяжении медианных 35,2 месяца (26,8–48,9) результаты получились следующими [1] [2] [3].
Общая выживаемость в группе «Опдиво» с «Ервоем» вышла к 23,7 месяца (95% ДИ [здесь и далее]: 18,8–29,4) — против 20,6 месяца (17,5–22,5) в группе «Ленвимы» или «Нексавара». Риск смерти снизился на относительный 21%: отношение риска (hazard ratio, HR) 0,79 (0,65–0,96); p=0,018.
Вероятность остаться в живых на протяжении 24 месяцев составила 49% против 39%, 36 месяцев — 38% против 24%.
Частота общего ответа (ORR) составила 36% (31–42), включая 7% полных ответов (CR), — против 13% (10–17), в том числе 2% CR (p<0,0001).
Медиана длительности ответа (DoR) получилась равной 30,4 месяца (21,2–NE) — против 12,9 месяца (10,2–31,2).
Медиана выживаемости без прогрессирования (PFS) обозначилась на уровне 9,1 месяца (6,6–10,5) — против 9,2 месяца (7,9–11,1). Риск прогрессирования заболевания или смерти снизился на относительных 13%: HR 0,87 (0,72–1,06). Статус PFS в течение 18 месяцев оказался справедливым для 34% пациентов против 18%, 24 месяцев — 28% против 12%.
Назначение иммуноонкологического коктейля привело к снижению риска ухудшения симптомов заболевания на относительных 24%: HR 0,76 (0,62–0,93); p=0,0059.
КОНТРАРГУМЕНТЫ
Использование ленватиниба или сорафениба в качестве контрольной группы — сомнительный выбор «Бристол-Майерс Сквибб». Разумнее было остановиться на более эффективных схемах, представленных либо сочетанием атезолизумаба (atezolizumab) с бевацизумабом (bevacizumab), за которым стоит «Рош», либо дуэтом «Имфинзи» (Imfinzi, дурвалумаб) с «Имджудо» (Imjudo, тремелимумаб) — блокатора PD-L1 с блокатором CTLA-4, продвигаемым «АстраЗенека» (AstraZeneca). Первая схема является предпочтительной, вторая выступает альтернативной в случае противопоказаний к назначению бевацизумаба или его непереносимости.
Согласно нынешним рекомендациям Американского общества клинической онкологии (ASCO), атезолизумаб с бевацизумабом лидирует по эффективности первоочередного лечения гепатоцеллюлярной карциномы, улучшая выживаемость и сдерживая прогрессирование заболевания относительно сорафениба: OS HR 0,66 (0,52–0,85) и PFS HR 0,65 (0,53–0,81) [1] [2].
Применение дурвалумаба (durvalumab) с тремелимумабом (tremelimumab) характеризуется только улучшением выживаемости относительно сорафениба: OS HR 0,78 (0,67–0,92) и PFS HR 0,90 (0,77–1,05) [3] [4].
ЭКСПЕРТНЫЕ КОММЕНТАРИИ
Начиная с марта 2020 года, «Опдиво» с «Ервоем» применяются во второлинейной терапии гепатоцеллюлярной карциномы. Но бизнес «Бристол-Майерс Сквибб» требует расширения охвата пригодных пациентов.
Результаты клинической проверки этого сочетания в перволинейной терапии рака печени получились на первый взгляд идентичными таковым в случае перволинейного использования дурвалумаба с тремелимумабом. Но есть важные отличия.
Во-первых, «Опдиво» с «Ервоеем» обеспечили более продолжительную общую выживаемость с медианой 20,6 месяца — против 16,4 месяца в случае «Имфинзи» с «Имджудо».
Во-вторых, они повысили шансы остаться в живых: до 49% и 38% на протяжении 24 и 36 месяцев — против 41% и 31%.
В-третьих, на лечение ответила большая пропорция пациентов: ORR 36% и CR 7% — против ORR 20% и CR 3%.
В-четвертых, ответ на лечение продолжался дольше: 30,4 месяца — против 22,3 месяца.
Однако справиться с прогрессированием заболевания удалось аналогично плохо.
Осталось разобраться с нежелательными явлениями и их частотой, чтобы понять, насколько новая схема лечения окажется переносимой.
Перспективность ниволумаба с ипилимумабом в контексте первоочередного лечения гепатоцеллюлярной карциномы сомнений не вызывает, ведь долгосрочные 4-летние наблюдения за пациентами, получавшими дурвалумаб с тремелимумабом, установили прилично высокую пропорцию долгожителей, то есть перешагнувших отметку 36-месячной выживаемости: каждый четвертый (25%) — против 15% в группе сорафениба [1].
В идеале было бы правильным напрямую сравнить эти две схемы с «Тецентриком» и «Авастином», но гранды фармотрасли на такое вряд ли пойдут.
ОДНАКО
Списывать сорафениб или ленватиниб со счетов пока рано. Согласно систематическому обзору и метаанализу, сочетание тирозинкиназного ингибитора, блокатора PD-1 и локорегионарной терапии характеризовалось максимальными шансами на конверсию изначально неоперабельной гепатоцеллюлярной карциномы в поддающуюся радикальной резекции, то есть с потенциалом очень длительной выживаемости [1].
Среднестадийная клиническая проверка подтвердила, что экспериментальное однотаблеточное сочетание биктегравира (bictegravir) с ленакапавиром (lenacapavir), принимаемое один раз в день, успешно справляется с задачей вирусной супрессии.
Терапия характеризовалась приемлемой переносимостью. Невыраженные и относительно нечастые побочные эффекты всё же имели место быть. Их появление связано, главным образом, с переключением со стабильной АРТ-схемы на новую.
Новая комбинация позволит ВИЧ-инфицированным наконец-то перейти к куда более удобному способу лечения либо извлечь дополнительную пользу, если их нынешняя комплексная АРТ-схема недостаточно эффективна.
Gilead Sciences создала абсолютную защиту от заражения ВИЧ двумя инъекциями в год. Ленакапавир: самый полный обзор в мире.
ПРЯМАЯ РЕЧЬ
«Одним из самых многообещающих аспектов исследования является то, что большинство участников (72%) принимали усиленный ингибитор протеазы — и смогли отказаться от него, перейдя на комбинированную терапию. Это мгновенно устраняет ряд лекарственных взаимодействий и снимает необходимость в обязательном приеме пищи».
Элизабет Шерман (Elizabeth Sherman), специалист по клинической фармации в области ВИЧ/СПИДа из Университета Нова Саутистерн (шт. Флорида, США).
СУТЬ ВОПРОСА
Хотя однотаблеточные АРТ-схемы, комбинирующие несколько препаратов в одной таблетке, уже больше десяти лет являются мировым стандартом, приблизительно 8% людей, живущих с ВИЧ, не могут им следовать по ряду причин, таких как резистентность, непереносимость, токсичность, лекарственные взаимодействия или противопоказания [1] [2] [3] [4].
Таким людям приходится обращаться к комплексным АРТ-схемам, которые представлены следующими вариантами:
схема, содержащая либо усиленный ингибитор протеазы (PI), либо ненуклеозидный ингибитор обратной транскриптазы (NNRTI) — вместе с одним (или больше) препаратом из класса, отличного от нуклеозидных или нуклеотидных ингибиторов обратной транскриптазы (NRTI, NtRTI);
схема, содержащая две (или больше) таблетки или требующая дозирования более одного раза в день;
схема, содержащая одновременно инъекционные лекарства (за исключением полноценной схемы длительного действия) и пероральные препараты.
БОЛЬШИЕ ЧИСЛА
Мировой фармацевтический рынок предлагает 11 однотаблеточных вариантов лечения ВИЧ-инфекции — клинический опыт показывает, что и этого недостаточно.
КАК ЭТО РАБОТАЕТ
Биктегравир (bictegravir; BIC) представляет собой ингибитор переноса цепи интегразой (INSTI) с высоким барьером для развития резистентности [1]. Ленакапавир (lenacapavir; LEN) — первый представитель ингибиторов капсида ВИЧ, к которому изначально нет резистентности независимо от предшествовавшего лечения [2].
Ленакапавир примкнул к ибализумабу и фостемсавиру в борьбе с резистентным вирусом иммунодефицита человека.
КЛИНИЧЕСКИЕ ПОДРОБНОСТИ
Клиническое исследование ARTISTRY-1 (NCT05502341) фазы II/III (рандомизированное, открытое, с активным контролем, многоцентровое, международное), охватившее находящихся в статусе вирусной супрессии (вирусная нагрузка РНК ВИЧ-1 < 50 копий/мл или неопределяемая) взрослых людей (n=128) с ВИЧ-1, проверило оправданность переключения с комплексной АРТ-схемы на экспериментальную ежедневную пероральную комбинацию, составленную из биктегравира (75 мг; BIC) и ленакапавира (25 или 50 мг; LEN).
Среди основных характеристик участников:
возраст: медианных 60 лет (26–27);
мужчин: 81%;
число предшествовавших АРТ-схем: медианных 6 (3–11);
продолжительность ВИЧ-терапии: медианных 27 лет (19,9–32,0);
число T-лимфоцитов CD4+: медианных 610 клеток/мкл (435–766);
наличие мутаций резистентности к INSTI, NNRTI, NRTI или PI: у 0%, 52%, 64% и 36% соответственно;
основная причина выбора лечения ВИЧ комплексной АРТ-схемой: резистентность (у 81%), непереносимость (30%), противопоказания (9%);
Основные характеристики комплексной АРТ-схемы участников:
число таблеток в день: медианных 3 (2–9);
число АРТ-классов: медианных 2 (1–5);
частота приема АРТ-препаратов: ежедневно (90% участников), дважды в день (41%), иной режим (1%).
Испытуемых либо переводили на экспериментальную АРТ-схему, либо оставляли следовать прежним курсом комплексной АРТ-схемы.
По прошествии 24 недель к первичной конечной точке эффективности лечения, заявленной пропорцией пациентов, продемонстрировавших вирусную нагрузку РНК ВИЧ-1 ≥ 50 копий/мл, то есть столкнувшихся с вирусологической неудачей, вышли 0,0% (n=0/51), 1,9% (n=1/52) и 0,0% (n=0/25) участников в группах BIC + LEN 25 мг, BIC + LEN 50 мг и контроля. Впрочем, впоследствии, этот один человек с вирусологическим провалом вернулся к статусу вирусной супрессии [1] [2].
Вирусная супрессия оказалась справедливой для 96,1% (n=49/51), 96,2% (т=50/52) и 100,0% (n=25/25) участников.
Усредненное изменение числа T-лимфоцитов CD4+ было следующим: +33 (p=0,9893), +5 (p=0,7173) и +43 клеток/мкл.
Назначение BIC + LEN характеризовалось приемлемой переносимостью. Среди наиболее распространенных нежелательных явлений, связанных с лечением (их частота не превышала 10%): диарея, гипертония, запор, кашель, назофарингит.
По истечении 48 недель терапии вирусологическая неудача была отмечена для 3,9% (n=2/51), 1,9% (n=1/52) и 0,0% (n=0/25) испытуемых [3].
Статус вирусной супрессии был подтвержден для 92,2% (n=47/51), 90,4% (n=47/52) и 100% (n=25/25) пациентов.
Следует понимать, что полученные сведения нельзя назвать полными ввиду отсутствия вирусологических данных у 2 и 4 участников в группах BIC + LEN 25 мг и BIC + LEN 50 мг.
Изменение числа T-лимфоцитов CD4+ было сопоставимым во всех трех группах.
Профиль безопасности BIC + LEN каких-либо опасений не вызывал.
КОНТРАРГУМЕНТЫ
Клиническому изучению биктегравира с ленакапавиром не помешала бы дополнительная информация, имеющая отношение к известным мутациям антиретровирусной резистентности, фармакокинетике, потенциальным лекарственным взаимодействиям.
Более продолжительные исследования ответят на вопрос, как долго люди, следующие новым АРТ-курсом, смогут поддерживать вирусную супрессию.
Пока точно неизвестно, насколько хорошо биктегравир с ленакапавиром способен справиться с АРТ-задачами в случае, во-первых, пациентов без исходной вирусологической супрессии, которые уже принимают множество АРТ-препаратов, и, во-вторых, людей с ВИЧ, которые прежде не лечились.
Нельзя также забывать о проблеме коинфекции ВИЧ и вируса гепатита B: похоже, все однотаблеточные схемы и препараты длительного действия, включая находящиеся на поздних стадиях разработки, не отвечают потребностям данной категории пациентов.
ЧТО ДАЛЬШЕ
В 48-недельной фазе III данного клинического испытания, которое завершится к концу 2025 года, осуществляется проверка экспериментального однотаблеточного сочетания BIC + LEN 50 мг. Выбор в пользу повышенной дозы ленакапавира обусловлен желанием упростить жизнь людей, живущих с ВИЧ: даже если человек случайно пропустит прием препарата, более высокая доза как бы «простит» этот промах. Простота, когда речь идет о лечении ВИЧ, равнозначна приверженности.
Эта же комбинация тестируется в клиническом исследовании ARTISTRY-2 (NCT06333808) фазы III, участников которого переводят со стабильной терапии препаратом «Биктарви» (Biktarvy, биктегравир + эмтрицитабин + тенофовира алафенамида фумарат; BIC/FTC/TAF).
Посттравматическое стрессовое расстройство (ПТСР) — серьезное и опасное для жизни нервно-психическое заболевание.
С ПТСР сталкивается большое количество людей.
Все нынешние методы лечения ПТСР характеризуются явно недостаточной эффективностью.
Разработан препарат для лечения ПТСР, который, располагая совершенно новым механизмом действия и не будучи психоактивным соединением, характеризуется высокой эффективностью, превосходящей селективные ингибиторы обратного захвата серотонина (СИОЗС), такие как сертралин (sertraline) и пароксетин (paroxetine).
У экспериментального лекарства нет серьезных побочных эффектов.
ЧТО ПРОИЗОШЛО
Австралийская «Байономикс» (Bionomics) справилась со среднестадийной клинической проверкой экспериментального анксиолитика BNC210, предназначенного для лечения посттравматического стрессового расстройства (ПТСР).
Терапевтическая эффективность BNC210 значительно превзошла таковую, предоставляемую одобренными лекарствами против ПТСР, притом что существующие препараты не обеспечивают должного выхода к ремиссии для большинства пациентов.
На момент анонса, состоявшегося в конце сентября 2023 года, Уолл-стрит отреагировал ростом биржевых котировок «Байономикс» сразу на 400%. Впрочем, впоследствии курс акций предприятия плавно снизился до исходного уровня.
«Байономикс» продолжает подготовку к запуску регистрационной клинической программы. Если BNC210 получит регуляторное одобрение, он станет первым новым за минувшие два десятка лет лекарством для терапии посттравматического стрессового расстройства.
Препарат-кандидат BNC210 представляет собой первый в своем классе отрицательный аллостерический модулятор никотинового ацетилхолинового рецептора α7, разрабатываемый для лечения тревожных, травматических или стрессовых расстройств.
Параллельно «Байономикс» изучает анксиолитик BNC210 в неотложном лечении социального тревожного расстройства (социофобии), и здесь он может стать первым лекарством, не вызывающим седативного эффекта и привыкания.
ПОЧЕМУ ЭТО ВАЖНО
Посттравматическое стрессовое расстройство (ПТСР) — серьезное нервно-психическое заболевание, с которым ежегодно сталкивается большое количество людей: к примеру, в США и Европе распространенность ПТСР в течение жизни составляет 6,4–8,0% [1] [2] [3].
Особенно широко ПТСР распространено среди участников или свидетелей боевых действий. Тем не менее подавляющее большинство (80% пациентов) с этим диагнозом представлено гражданской популяцией, не имеющей никакого отношения к военным конфликтам [12] [14].
ПТСР — серьезное и опасное для жизни патологическое состояние, ассоциированное с повышенной смертностью, кардиометаболической заболеваемостью и риском суицида. ПТСР отрицательно влияет на повседневную жизнь человека, часто приводит к разрыву отношений, депрессии, снижению повседневной активности, ухудшению когнитивных и психосоциальных функций, злоупотреблению психоактивными веществами и высокозатратному использованию медицинских услуг.
Лечение ПТСР особенно сложно в таких нередких случаях, как диссоциативный подтип ПТСР, повторное воздействие травмы, наличие сопутствующих заболеваний, включая аффективные расстройства и расстройства, связанные с употреблением алкоголя и психоактивных веществ [4] [5] [6].
Комплексная природа ПТСР ассоциирована с обострением симптомов, резистентностью к лечению и прекращением терапии [5] [7]. Психотерапия, ориентированная на травму, является золотым стандартом лечения ПТСР, однако у многих людей симптоматика сохраняется, а частота отказа от лечения высока [8] [9] [10]. Фармакотерапия ПТСР, предполагающая прием селективных ингибиторов обратного захвата серотонина (СИОЗС), не обеспечивает должного эффекта у приблизительно 35–47% пациентов [11].
Для решения проблемы индивидуального, общественного и экономического бремени ПТСР необходимы более эффективные терапевтические подходы [12] [13].
ЧТО ВЫЯСНИЛОСЬ
Клиническое исследование ATTUNE (NCT04951076) фазы IIb (рандомизированное, двойное слепое, плацебо-контролируемое, многоцентровое, международное) пригласило взрослых пациентов (n=212) с диагнозом посттравматического стрессового расстройства (ПТСР).
Среди основных требований к участию: балл выраженности симптомов ≥ 30 по шкале клинической оценки ПТСР по DSM-5 (CAPS-5); индексное травматизирующее событие произошло во взрослом возрасте.
Среди базовых характеристик пациентов: женщин 64%; средний возраст 42 года (19–68); средний балл CAPS-5 41,5 (30–59).
Испытание включило людей с обширным спектром причин, послуживших развитию ПТСР, как то: физическое или сексуальное насилие, внезапная насильственная смерть, нападение с применением оружия, участие или пребывание в зоне боевых действий, дорожно-транспортное происшествие, опасное для жизни заболевание или травма, внезапная смерть от несчастного случая, пребывание в плену, пожар или взрыв, причиненные другому человеку серьезные травмы, вред здоровью или смерть, серьезный несчастный случай на работе, дома или во время активного отдыха, тяжелые человеческие страдания, иной нежелательный или дискомфортный сексуальный опыт, иное очень стрессовое событие или переживание.
На протяжении 12 недель пациенты получали перорально плацебо или BNC210 в дозе 900 мг дважды в день.
Назначение BNC210 обеспечило выход к первичной конечной точке эффективности лечения, установленной изменением общего балла тяжести симптомов ПТСР по шкале CAPS-5: усредненная разница с плацебо составила −4,03 пункта (p=0,048) [1] [2].
Статистически значимая разница с плацебо проявилась даже раньше: на 4-й (p=0,015) и 8-й неделях (p=0,014) терапии: соответственно −4,11 и −4,74 пункта.
Применение BNC210 обеспечило улучшение симптомов, особенно сложных для купирования при ПТСР: симптомы интрузии (критерий B) и негативные мысли и эмоции (критерий D). Так, на 4-й, 8-й и 12-й неделях лечения усредненная разница с плацебо балла интрузии составила −0,98 (p=0,046), −1,35 (p=0,035) и −1,33 (p=0,047) пункта, а балла негативных мыслей и эмоций — −2,05 (p=0,005), −1,79 (p=0,036) и −1,79 (p=0,061) пункта.
Назначение BNC210 привело к существенному улучшению вторичных конечных точек. В частности, на 12-й неделе лечения было отмечено облегчение симптомов депрессии и сна, согласно шкале Монтгомери — Осберг для оценки депрессии (MADRS) и индексу тяжести бессонницы (ISI) соответственно: усредненная разница с плацебо составила −3,19 (p=0,040) и −2,19 (p=0,041) пункта.
Наблюдалась положительная динамика в изменении других показателей, в том числе оцениваемых по шкале общего клинического впечатления о тяжести заболевания (CGI-S), по шкале общего впечатления пациента о тяжести заболевания (PGI-S), по шкале Гамильтона для оценки выраженности тревоги (HAM-A), по шкале инвалидизации Шихана (SDS).
Лечение посттравматического стрессового расстройства при помощи BNC210 характеризовалось приемлемыми переносимостью и безопасностью, тем самым указывая на возможность хронического применения этого препарата.
Среди наиболее распространенных нежелательных явлений (НЯ) при назначении BNC210: головная боль (у 17% пациентов против 13% в группе плацебо), тошнота (12% против 8%), усталость (6% против 8%) — в подавляющем большинстве случаев они носили легко-умеренную степень выраженности.
По сравнению с плацебо не зафиксировано избыточных психоактивных НЯ, таких как седация, привыкание и когнитивные нарушения, что выгодно отличает BNC210 от существующего арсенала лекарственных препаратов против ПТСР. Применение BNC210 не вызывало НЯ сексуального характера, таких как снижение либидо и эректильная дисфункция.
Был отмечен рост уровня печеночных ферментов (у 13% пациентов против 2% в группе плацебо), хотя это не было связано с функциональными нарушениями печени (то есть не было ее лекарственного поражения, декомпенсации печеночного заболевания, проявления закона Хая, повышения уровня биллирубина), протекало бессимптомно и в большинстве случаев разрешалось без необходимости в отмене лечения.
Массив научных данных указывает на участие холинергической системы в этиологии и лечении поведения, связанного со страхом. Никотиновые ацетилхолиновые рецепторы (nAChR) — это ионотропные лиганд-связанные рецепторные полипептиды, активируемые ацетилхолином и никотином [2] [3]. Подтип α7 состоит из пяти субъединиц α7, которые в основном гомомерны [4].
В целом NAChR являются неселективными катионными каналами, причем подтип α7 особенно проницаем для кальция [5], а также для натрия [6]. Исследования на крысах выявили роль α7 nAChR в анксиолитическом поведении. Антагонизм α7 nAChR, концентрация которого высока в миндалевидном теле крыс [7], отразился на поведении, связанного со страхом [8].
Селективные ингибиторы обратного захвата серотонина (СИОЗС), как было показано, ингибируют никотиновые ацетилхолиновые рецепторы, то есть СИОЗС, как предполагается, действуют через холинергическую, а не серотонинергическую модуляцию [9]. Хотя это не было продемонстрировано на людях, крысы засвидетельствовали холинергическое торможение активности миндалевидного тела после приема бензодиазепинов [10]. Это оставляет вероятность для того, что лечение тревоги бензодиазепинами действует, не исключено, через холинергическое торможение в дополнение к ГАМК-ергическим эффектам.
На базе доклинических моделей о роли холинергической системы в поведении, связанном со страхом, высказано предположение, что модуляция α7 nAChR может служить анксиолитиком. С этой целью был разработан BNC210.
В доклинических моделях BNC210 хорошо переносился, был безопасен и не вызывал седативного эффекта [11]. BNC210 эффективно работал как анксиолитик в различных поведенческих парадигмах, в том числе превзошел плацебо в тестах на тревожность: темно-светлая камера (для мышей) и приподнятый крестообразный лабиринт (для крыс) [3]. Изучение профиля безопасности и переносимости BNC210 в исследованиях на людях выявило обнадеживающие результаты без седативных эффектов, присущих типичным анксиолитикам [12] [13].
ЧТО ДАЛЬШЕ
«Байономикс» (Bionomics) в четвертом квартале 2024 года, после консультации с Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA), намеревается организовать опорное клиническое испытание фазы III, которое окончательно установит эффективность и безопасность экспериментального BNC210 для лечения посттравматического стрессового расстройства (ПТСР). Результаты будут собраны в 2025–2026 гг.
Ввиду определенного негативного влияния на печеночные ферменты будет также протестирована сниженная доза BNC210.
ПЕРСПЕКТИВЫ
Как утверждает «Байономикс» (Bionomics), эффект лечения и размер эффекта, обеспечиваемые экспериментальным анксиолитиком BNC210 в ходе лечения посттравматического стрессового расстройства (ПТСР), соответственно превосходят и сравнимы с таковыми при применении селективных ингибиторов обратного захвата серотонина (СИОЗС).
Так, BNC210, согласно шкале клинической оценки ПТСР по DSM-5 (CAPS-5), обеспечил снижение общего балла тяжести симптомов на абсолютных 20 пунктов, тогда как сертралин (sertraline) снизил его на на абсолютных 6,8 и 9,8 пункта (в разных клинических испытаниях), а пароксетин (paroxetine) — на 14 и 11 пункта [1].
Размер эффекта при назначении BNC210 составил 0,40; 0,44 и 0,40 — соответственно после 4, 8 и 12 недель лечения [2].
Лечение при помощи BNC210 прекратили 20% испытуемых — против 10% в контрольной группе, что, впрочем, предсказуемо ввиду нестабильного поведения пациентов с ПТСР.
Прямым конкурентом BNC210 выступает экспериментальная MDMA-психотерапия, в двух клинических исследованиях фазы III, MAPP1 (NCT03537014) и MAPP2 (NCT04077437), продемонстрировавшая снижение общего балла CAPS-5 на абсолютных 26,2 и 23,7 пункта, притом что соответствующий размер эффекта (относительно плацебо) составил 0,91 и 0,7 [3] [4].
Для сравнения: размер эффекта при назначении сертралина и пароксетина изрядно уступает обеспечиваемому MDMA-психотерапией, будучи втрое и вдвое меньше — 0,31–0,37 и 0,45–0,56. Кроме того, если назначение этих СИОЗС сопровождалось высокой частотой выбывания пациентов из клинических испытаний (отказ от лечения на уровне 28–39%), то при MDMA-психотерапии пациенты вообще не стремились ее прекратить.
В ходе MDMA-психотерапии на лечение ответили 80–87% пациентов (клинически значимое снижение общего балла тяжести симптомов ПТСР по шкале CAPS-5 минимум на 10 пунктов), потеряли диагноз 67–71% испытуемых (статус респондента с одновременным прекращением соответствия диагностическим критериям ПТСР), к ремиссии вышли 33–46% человек (потеря диагноза и общий балл CAPS-5 менее 11 пунктов).
Несмотря на приличную результативность MDMA-психотерапии в лечении ПТСР, она завязана на применении психоделического (эмпатогенного) вещества, и потому ее распространение в мире ожидается весьма ограниченным ввиду строгости законодательства в отношении психоактивных соединений, пусть даже используемых исключительно в медицинских целях, поскольку они обладают высоким потенциалом злоупотребления.
Для понимания дальнейших перспектив BNC210 в лечении ПТСР, следует обратить внимание на такие важные для врачей и пациентов показатели, как размер эффекта и степень приверженности лечению, а также пропорции ответивших на лечение пациентов, потерявших диагноз и вышедших к ремиссии. Сведения об указанных пропорциях «Байономикс» пока не раскрыла.
Как бы то ни было, получилось, что терапевтическая эффективность анксиолитика BNC210 сравнима с таковой у MDMA-психотерапии, если речь идет о достаточно тяжелом ПТСР (общий балл тяжести симптомов CAPS-5 ≥ 40).
Существующие лекарства для снижения артериального давления недостаточно эффективны, со временем к ним развивается привыкание.
Блокирование выработки ангиотензиногена в печени — решительно новый действенный способ удержания давления в норме.
Предполагается очень редкий режим дозирования грядущих лекарств: один раз в месяц, квартал или даже полгода.
Зилебесиран (zilebesiran) и тонламарсен (tonlamarsen) — быстро, надежно, дорого!
ЧТО ПРОИЗОШЛО
«Алнайлам фармасьютикалс» (Alnylam Pharmaceuticals) и «Айонис фармасьютикалс» (Ionis Pharmaceuticals) разработали новаторские препараты, предназначенные для лечения гипертонии.
Передовое лечение обещает помочь даже в том случае, если артериальное давление никак не поддается снижению имеющимся арсеналом гипотензивных лекарственных средств.
Экспериментальные зилебесиран (zilebesiran) и тонламарсен (tonlamarsen) обращаются к совершенно новому механизму действия, прежде не задействовавшемуся в лечении гипертонии и предполагающему подавление выработки ангиотензиногена в печени.
Существуют весомые предпосылки, что должная терапевтическая эффективность грядущих лекарств сохранится при очень редком режиме дозирования, когда препараты назначаются подкожными инъекциями один раз в три или даже шесть месяцев.
В дальнейшем эти лекарства могут превратиться в еще более удобные пероральные таблетки или капсулы, принимаемые, к примеру, один раз в неделю или месяц.
ПОЧЕМУ ЭТО ВАЖНО
Артериальная гипертензия, или гипертония, — сложное многофакторное заболевание, проявляющееся повышенным артериальным давлением крови.
Клинический диагноз гипертонии, согласно большинству консенсусных руководств, ставится, когда систолическое артериальное давление превышает 140 мм рт. ст., а диастолическое — 90 мм рт. ст. [1] [2] [3]. При этом, однако, Американская кардиологическая ассоциация (AHA) совместно с Американской коллегией кардиологов (ACC) рекомендуют ставить диагноз артериальной гипертензии при соответствующих показателях выше 130 и 80 мм рт. ст. [4].
Гипертонией страдает каждый четвертый мужчина и каждая пятая женщина на планете — всего свыше 1,28 млрд жителей Земли в возрасте 30–79 лет [5].
Гипертония — одна из ведущих причин преждевременной смерти.
Неконтролируемая гипертония, то есть когда ее терапия не проводится надлежащим образом или она вообще остается без какого-либо лечения, увеличивает риск развития или ухудшения сердечно-сосудистых заболеваний и нарушений мозгового кровообращения — ишемической болезни сердца и инсульта, а также способствует прогрессированию хронической болезни почек [6] [7] [8] [9].
Несмотря на наличие эффективных лекарственных препаратов для снижения давления, фармакотерапия гипертонии оставляет желать много лучшего. При этом половине больных, придерживающихся хронического приема гипотензивных лекарственных средств, не удается выйти к целевым показателям артериального давления [10].
Частично это является следствием, во-первых, неспособности лечащего врача начать или интенсифицировать (усилить) медикаментозную терапию и, во-вторых, плохой приверженности пациентов к ежедневному приему пероральных лекарственных препаратов [11] [12] [13].
Ситуация осложняется тем, что даже когда артериальное давление кажется хорошо контролируемым на основе периодических измерений, контроль над гипертонией может оставаться неоптимальным из-за выраженной вариабельности артериального давления в течение суточного цикла и в долгосрочной перспективе [14] [15] [16].
РЕШЕНИЕ ПРОБЛЕМЫ
Хроническая гиперактивность ренин-ангиотензин-альдостероновой системы (РААС) считается одним из основных факторов патогенеза сердечно-сосудистых заболеваний, включая гипертонию, сердечную недостаточность, сердечную гипертрофию, атеросклероз [1] [2].
Ингибирование сигнального пути РААС ингибиторами ангиотензинпревращающего фермента (АПФ) или блокаторами рецепторов ангиотензина II (БРА) — один из наиболее эффективных методов лечения артериальной гипертензии и сердечной недостаточности со сниженной фракцией выброса (HFrEF) [3] [4] [5] [6] [7].
Однако ключевым препятствием для реализации этого подхода в полную силу является то, что он увеличивает риск целевой токсичности, проявляющейся главным образом гиперкалиемией и нарушением функции почек. Другими словами, приходится прибегать к более низким клиническим дозам, что тем самым ограничивает возможность для оптимального ингибирования РААС, которое иначе предоставило бы более выраженные клинические преимущества в отношении снижения артериального давления [8].
Фармакологическое ингибирование нисходящего каскада сигнального пути РААС также приводит к формированию компенсаторных путей в вышележащем каскаде, что еще больше снижает терапевтическую эффективность гипотензивных лекарственных препаратов [9] [10] [11].
Нацеливание на верхний каскад сигнального пути РААС путем сайленсинга (подавления экспресии) вырабатываемого печенью ангиотензиногена (AGT) — принципиально новый подход к сдерживанию активности РААС [12].
Поскольку ангиотензиноген является единственным предшественником всех пептидов ангиотензина, его сайленсинг характеризуется рядом потенциальных преимуществ по сравнению с большинством существующих способов ингибирования РААС [13] [14] [15] [16] [17] [18].
Так, тканеспецифическое подавление выработки ангиотензиногена в печени, никак не затрагивающее ингибирование РААС в почках, позволяет надеяться на улучшенный профиль безопасности, потому что сохраняются почечный гомеостаз и механизм обратной канальцево-клубочковой связи. В итоге снижаются риски роста уровня калия и почечной дисфункции.
К минимуму сведен риск развития компенсаторных механизмов, которые проявляются в ответ на применение АПФ или БРА и которые парадоксальным образом пытаются восстановить уровни ренина, альдостерона и ангиотензина II.
Более полное ингибирование локальной выработки ангиотензина II в сосудистой или сердечной тканях принесет пользу пациентам с резистентной гипертонией или сердечной недостаточностью.
ALNYLAM PHARMACEUTICALS И НОВАТОРСКОЕ ЛЕЧЕНИЕ ГИПЕРТОНИИ
«Алнайлам фармасьютикалс» (Alnylam Pharmaceuticals) разрабатывает экспериментальный препарат зилебесиран (zilebesiran), построенный на терапевтической модальности РНК-интерференции, неоднократно доказавшей собственную медицинскую состоятельность.
Раннестадийная клиническая проверка зилебесирана оказалась весьма успешной, и потому этот препарат-кандидат приглянулся «Рош» (Roche), которая в конце июля 2024 года оформила с «Алнайлам» партнерское соглашение [1].
Согласно договоренностям, швейцарский фармацевтический гигант выдаст «Алнайлам» авансом 310 млн долларов, а затем будет выплачивать дополнительные суммы по мере разработки, регуляторного одобрения и продаж готового препарата — совокупно до 2,8 млрд долларов. Реализация зилебесирана в США будет осуществляться совместно на равных условиях извлечения прибыли, во всех других странах продажей будет заниматься «Рош», отчисляя «Алнайлам» определенное роялти.
ЗИЛЕБЕСИРАН: КАК ОН РАБОТАЕТ
Ренин-ангиотензин-альдостероновая система (РААС) представляет собой сигнальный каскад, который надежно доказал свою определяющую роль в регуляции артериального давление и подавление которого сопровождается хорошо известными гипотензивными эффектами [1] [2] [3] [4] [5] [6] [7].
Вопрос о том, блокировать или не блокировать РААС, давно не является актуальным в клинических сценариях сердечной недостаточности, диабетической нефропатии, хронической болезни почек. Усилия фармотрасли направлены на оптимизацию этой блокады.
[membership level=»2,3″ show_noaccess=»true»]
Зилебесиран (zilebesiran, ALN-AGT) таргетирован против ангиотензиногена (AGT), который является самым ранним предшественником ангиотензина I (проангиотензина), дающего начало ангиотензину II — олигопептидному гормону, который входит в состав РААС и который вызывает сужение сосудов и повышение артериального давления [8].
Зилебесиран построен на базе РНК-интерференции (RNAi) — естественного механизма сайленсинга генов (подавления их экспрессии) на стадии транскрипции, трансляции, деаденилирования или деградации матричной РНК (мРНК) при помощи малой интерферирующей РНК (миРНК). За открытие RNAi была вручена Нобелевская премия в 2006 году [9].
Зилебесиран вмешивается в работу мРНК гена AGT, что отражается его сайленсингом. Ингибирование синтеза ангиотензиногена в печени приводит к устойчивому снижению уровня ангиотензина I и в конечном итоге ангиотензина II. В следствии этого снижается артериальное давление, поскольку ангиотензин II равно как самостоятельно оказывает сосудосуживающий эффект, так и делает это посредством стимуляции синтеза вазопрессина.
Конструктивно зилебесиран состоит из двух частей: химически модифицированной миРНК против AGT и N-ацетилгалактозаминового (GalNAc) лиганда, связывающего асиалогликопротеиновый рецептор 1 (ASGR1) на гепатоцитах в целях таргетной доставки препарата в печень, где эндосомальное накопление миРНК, медленная их утилизация и рециклинг подпитывают нокдаун AGT на протяжении длительного времени.
Зилебесиран обращается к фирменной технологии GalNAc-конъюгатов Enhanced Stabilization Chemistry Plus (ESC+), которая позволяет осуществлять весьма редкое подкожное введение лекарства, вкупе наделяя его высокой селективностью и широким терапевтическим индексом.
[/membership]
ЗИЛЕБЕСИРАН: ВСЁ ПОЛУЧИТСЯ
В пользу того, что технологический подход «Алнайлам фармасьютикалс» (Alnylam Pharmaceuticals) сработает в задаче лечения гипертонии, свидетельствуют поставленные ею на коммерческие рельсы РНК-интерференционные препараты против редких заболеваний:
«Онпаттро» (Onpattro, патисиран) и «Амвуттра» (Amvuttra, вутрисиран): лечение наследственного транстиретинового амилоидоза (hATTR) с полинейропатией (hATTR-PN), ранее известного как семейная амилоидная полинейропатия (FAP) [1] [2].
«Окслумо» (Oxlumo, лумасиран): лечение первичной гипероксалурии 1-го типа (PH1) [4] [5].
Все эти препараты применяются в режиме редкого дозирования. Так, «Онпаттро» требует внутривенных вливаний один раз в три недели, «Гивлаари» необходимо назначать подкожными инъекциями один раз в месяц, а «Окслумо» и «Амвуттра» вводятся подкожно один раз в квартал.
Кроме того, «Новартис» (Novartis) выпустила«Леквио» / «Сибрава» (Leqvio / Sibrava, инклисиран) — РНК-интерференционный препарат, предназначенный для мощного снижения уровня холестерина липопротеинов низкой плотности (ЛПНП) посредством подкожных инъекций один раз в полгода. За оригинальной идеей «Леквио» / «Сибрава» стоит всё та же «Алнайлам».
Две инъекции инклисирана в год — и о высоком холестерине можно забыть.
ЗИЛЕБЕСИРАН: ЭФФЕКТИВНОСТЬ И БЕЗОПАСНОСТЬ
NCT03934307
Клиническое исследование NCT03934307 фазы I (рандомизированное, двойное слепое, плацебо-контролируемое, многоцентровое) привлекло взрослых пациентов с повышенным систолическим давлением (легко-умеренная гипертония: 130–165 мм рт. ст.), не принимающих каких-либо гипотензивных препаратов (либо ранее не лечивших гипертонию, либо отказавшихся от таких лекарств).
[membership level=»2,3″ show_noaccess=»true»]
Участники получили по одной подкожной дозе зилебесирана (zilebesiran) в различных дозах или плацебо.
Результаты в 200-мг подгруппе зилебесирана по прошествии 8 недель следующие [1]:
Снижение сывороточного уровня ангиотензиногена (AGT) более чем на 90% сохранялось в течение 3 месяцев.
Усредненное снижение систолического артериального давления (САД) и диастолического артериального давления (ДАД) составило 11±2 и 8±1 мм рт. ст., причем у некоторых испытуемых максимальное снижение составило 19 и 12 мм рт. ст.
В подгруппах зилебесирана в дозе 100 мг и выше отмечено устойчивое снижение сывороточного уровня AGT более чем на 90%, при этом в 800-мг подгруппе указанное снижение составило 96–98%.
Назначение зилебесирана в дозе 100 мг и выше обеспечило снижение САД не менее чем на 10 мм рт. ст., при этом в 800-мг подгруппе снижение САД превысило 15 мм рт. ст.
После 24 недель продемонстрированы следующие исходы [3]:
На 3-й неделе наблюдений применение зилебесирана в дозе 100 мг и выше привело к снижению сывороточного уровня AGT более чем на 90%, которое сохранялось на протяжении 12 недель, причем в 800-мг подгруппе указанное снижение было устойчивым в течение 24 недель.
На 8-й неделе наблюдений назначение зилебесирана в дозе 200 мг и выше привело к снижению САД не менее чем на 10 мм рт. ст. и продолжило сохранять свое клинически значимое падение вплоть до 24-й недели.
На 24-й неделе наблюдений в 800-мг подгруппе зилебесирана снижение САД превысило 20 мм рт. ст., причем у 6 из 8 пациентов этого удалось добиться без каких-либо дополнительных гипотензивных препаратов.
Зилебесиран в дозе 200 мг и выше обеспечил стабильное и устойчивое снижение САД и ДАД в дневное и ночное время, что свидетельствует о возможности достижения тонического контроля над артериальным давлением в течение длительного периода.
Назначение зилебесирана характеризовалось приемлемой переносимостью, каких-либо серьезных нежелательных явлений (НЯ) в ответ на его назначение не зарегистрировано.
«Алнайлам фармасьютикалс» (Alnylam Pharmaceuticals) дополнительно изучила 800-мг дозу зилебесирана на фоне низкосолевой диеты или попутного ежедневного назначения 300 мг ирбесартана (irbesartan), блокатора рецепторов ангиотензина II (БРА).
Проверка зилебесирана при рационе с низким содержанием соли, рекомендованной гипертоникам, была необходима для выяснения переносимости лечения ввиду истощения объема жидкости в организме по причине потери натрия, вызванного низкосолевой диетой, что способно привести к гипотензивным событиям.
Добавление ирбесартана результировало дополнительным снижением САД и ДАД на 6,4 и 3,2 мм рт. ст., причем без клинически значимых изменений сывороточных уровней креатинина и калия.
Выводы следующие: низкосолевая диета оказывает дополнительный эффект в виде усиленного снижения артериального давления, которое может упасть слишком низко, и потому в задаче предотвращения вероятных гипотензивных событий может пригодиться, напротив, высокосолевая диета.
«Алнайлам» также протестировала зилебесиран среди пациентов с гипертонией и ожирением II или III степени (индекс массы тела [ИМТ] > 35 кг/м2), но без сахарного диабета. Подтверждено выраженное и устойчивое снижение САД с отражающими механизм действия ростом уровня ренина и снижением уровней ангиотензина II и альдостерона.
[/membership]
KARDIA-1
Клиническое исследование KARDIA-1 (NCT04936035) фазы II (рандомизированное, двойное слепое, плацебо-контролируемое, многоцентровое) проверило мононазначение зилебесирана (zilebesiran) среди взрослых пациентов (n=394) с легко-умеренной гипертонией (усредненное дневное систолическое артериальное давление [САД] в пределах 135–160 мм рт. ст.), ранее не лечивших гипертонию либо следующих стабильным курсом гипотензивных препаратов (в любом случае испытуемые не принимали таковые в ходе исследования) [1].
[membership level=»2,3″ show_noaccess=»true»]
На протяжении 6-месячного двойного слепого периода участники получали либо зилебесиран в дозе 150, 300 или 600 мг каждые 6 месяцев или 300 мг каждые 3 месяца, либо плацебо каждые 3 месяца. В последующем 6-месячном открытом периоде пациенты из группы плацебо переводились на зилебесиран.
Изменения артериального давления (мм рт. см.) относительно плацебо фиксировались двумя способами: посредством суточного мониторирования артериального давления (СМАД) и однократного измерения артериального давления сфигмоманометром (ОИАДС).
По истечение 3 месяцев изменение САД, согласно СМАД, составило: −14,1 (95% ДИ [здесь и далее]: −19,2, −9,0), −16,7 (−21,2, −12,3) и −15,7 (−20,8, −10,6) — среди испытуемых, получивших зилебесиран в дозе 150 мг (каждые 6 месяцев), 300 мг (каждые 3 или 6 месяцев) и 600 мг (каждые 6 месяцев) [p<0,0001].
Изменение САД, согласно ОИАДС: −9,6 (−13,8, −5,3), −12,0 (−15,7, −8,3) и −9,1 (−13,4, −4,8) [p<0,0001].
По прошествии 6 месяцев изменение САД, согласно СМАД, получилось равным: −11,1 (−15,8, −6,4), −14,5 (−19,1, −9,9), −14,1 (−18,9, −9,4) и −14,2 (−18,9, −9,5) — среди участников, которым назначили зилебесиран в дозе 150 мг (каждые 6 месяц), 300 мг (каждые 6 месяцев), 300 мг (каждые 3 месяца) и 600 мг (каждые 6 месяцев) [p<0,0001].
Изменение САД, согласно ОИАДС: −7,5 (−12,4, −2,7), −10,5 (−15,3, −5,7), −12,1 (−17,2, −7,1) и −10,2 (−15,1, −5,3) [p=0,0025, p<0,0001].
Пропорции пациентов, ответивших на лечение, — то есть, согласно СМАД, достигших САД < 130 мм рт. ст. и/или его снижения на ≥ 20 мм рт. ст. без дополнительных гипотензивных лекарственных препаратов, — по прошествии 6 месяцев составили 7%, 31%, 51%, 39% и 47% — среди участников, получивших плацебо, зилебесиран в дозе 150 мг (каждые 6 месяц), 300 мг (каждые 6 месяцев), 300 мг (каждые 3 месяца) и 600 мг (каждые 6 месяцев). Соответствующие отношения шансов (odds ratio, OR) выйти к ответу для подгрупп зилебесирана составили 6,8 (2,4–19,4), 19,7 (6,8–56,9), 10,7 (3,8–30,6) и 17,9 (6,2–51,5) [p<0,0001].
Зилебесиран обеспечил непротиворечивое и устойчивое снижение артериального давления на протяжении всего суточного цикла, включая ночное время.
Назначение зилебесирана характеризовалось приемлемой переносимостью. Нежелательные явления (НЯ), связанные с гиперкалиемией (среди 5% пациентов, получавших зилебесиран, — против 1% в группе плацебо), носили легкую степень выраженности, были преходящими и разрешались самостоятельно. Число случаев гипотонии было небольшим (4% против 1%). Не отмечено клинических значимых изменений почечной или печеночной функций. Были зарегистрированы редкие случаи серьезных НЯ (4% против 7%) и тяжелых НЯ (3% против 4%).
Дополнительный анализ установил, что зилебесиран обеспечил должное снижение САД вне зависимости от пола, возраста, расы, а также исходных САД и расчетной скорости клубочковой фильтрации (eGFR). Разве что влияла исходная концентрация ренина в плазме у темнокожих пациентов: если она была низкой, то и снижение САД было не столь выраженным [5].
[/membership]
KARDIA-2
Клиническое исследование KARDIA-2 (NCT05103332) фазы II (рандомизированное, двойное слепое, плацебо-контролируемое, многоцентровое, международное) изучило добавление зилебесирана (zilebesiran) к гипотензивной терапии взрослых пациентов (n=800) с резистентной гипертонией, которая адекватно не контролируется лекарственными препаратами.
[membership level=»2,3″ show_noaccess=»true»]
Среди критериев включения: усредненное дневное систолическое артериальное давление (САД ) в пределах 155–180 мм рт. ст. или 145–180 мм рт. ст. (соответственно при ранее нелеченной гипертонии и леченной одним–двумя препаратами гипертонии), которое должно снизиться до 130–160 мм рт. ст. после вводного периода исследования.
Вводный 4-недельный открытый период, который был необходим для выравнивания исходных показателей САД, предполагал ежедневный однократный прием какого-либо одного гипотензивного лекарства: 2,5 мг диуретика индапамида (indapamide), 40 мг блокатора рецепторов ангиотензина II олмесартана (olmesartan) или 5 мг блокатора кальциевых каналов амлодипина (amlodipine) [1].
В последующем 6-месячном двойном слепом периоде участники однократно получили подкожную инъекцию 600 мг зилебесирана или плацебо — на фоне продолжения ежедневного приема ранее выбранного гипотензивного препарата (по прошествии 3 месяцев могли быть добавлены дополнительные лекарства, если гипертония не купировалась).
Изменения артериального давления (мм рт. см.) относительно плацебо фиксировались двумя способами: посредством суточного мониторирования артериального давления (СМАД) и однократного измерения артериального давления сфигмоманометром (ОИАДС).
По истечение 3 месяцев изменение САД, согласно СМАД, составило: −12,1 (95% ДИ [здесь и далее]: −16,5, −7,6), −9,7 (−12,9, −6,6) и −4,0 (−7,6, −0,3) — среди получивших зилебесиран испытуемых, придерживавшихся ежедневной гипотензивной терапии индапамидом, амлодипином или олмесартаном (p<0,001, p<0,001, p=0,036).
Изменение САД, согласно ОИАДС: −18,5 (−22,8, −14,2), −10,2 (−13,5, −6,9) и −7,0 (−10,4, −3,6) [p<0,001].
По прошествии 6 месяцев изменение САД, согласно СМАД, получилось равным: −11,0 (−14,7, −7,3), −7,9 (−10,6, −5,3) и −1,6 (−4,4, −1,2) [p<0,001, p<0,001, and p=0,26].
Изменение САД, согласно ОИАДС: −13,6 (−16,9, −10,3), −8,6 (−10,9, −6,3) и −4,6 (−6,8, −2,4) [p<0,001].
Пропорции пациентов, ответивших на лечение, — то есть, согласно СМАД, достигших САД < 130 мм рт. ст. и/или его снижения на ≥ 20 мм рт. ст. без дополнительных (к уже назначаемым) гипотензивных лекарственных препаратов, — по прошествии 6 месяцев составили 64%, 40% и 27% — среди участников, получивших зилебесиран, а затем следовавших терапии индапамидом, амлодипином или олмесартантом. В подгруппах плацебо пропорции таковы: 14%, 14% и 17%. Соответствующие отношения шансов (odds ratio, OR) выйти к ответу для подгрупп зилебесирана составили 12,4 (4,6–33,3), 5,1 (2,4–10,6), 1,8 (0,9–3,4) [p<0,001, p<0,001, p=0,077].
Назначение зилебесирана характеризовалось приемлемой переносимостью. Нежелательные явления (НЯ), в том числе лабораторного характера, носили легкую степень выраженности, были преходящими и разрешались самостоятельно. С гипотонией или ортостатической гипотонией столкнулись 0%, 6% и 5% пациентов, получивших зилебесиран, а затем придерживавших терапии индапамидом, амлодипином или олмесартаном, — против 0%, 3% и 3% в подгруппах плацебо.
[/membership]
KARDIA-3
Продолжается клиническое исследование KARDIA-3 (NCT06272487) фазы II (рандомизированное, двойное слепое, плацебо-контролируемое, многоцентровое), которое оценивает оправданность назначения зилебесирана (zilebesiran) в качестве дополнительного препарата среди пациентов (n=390) с высоким сердечно-сосудистым риском и резистентной гипертонией (систолическое артериальное давление 130–170 мм рт. ст.), которая адекватно не контролируется 2–4 классами гипотензивных лекарств с разными механизмами действия.
Среди основных критериев включения (на выбор): сердечно-сосудистое заболевание в анамнезе, высокий сердечно-сосудистый риск или хроническая болезнь почек (расчетная скорость клубочковой фильтрации [eGFR] 30–60 мл/мин/1,73 м2).
Исследование ожидается к завершению весной 2025 года.
ЗИЛЕБЕСИРАН: ОПТИМИСТИЧНЫЕ ПЕРСПЕКТИВЫ
Согласно консенсусному мнению, снижение артериального давления более чем на 5 мм рт. ст. является клинически значимым, и потому терапевтические показатели, обеспеченные зилебесираном (zilebesiran), выглядят весьма убедительными. Прогнозируемый однозначный коммерческий успех препарата обусловлен главным образом схемой его очень редкого дозирования (один раз в квартал или полгода), поскольку приверженность пациентов фармакотерапии гипертонии решительно недостаточна.
[membership level=»2,3″ show_noaccess=»true»]
Ввиду того, что зилебесиран селективно таргетирован против выработки ангиотензиногена в печени, оказываемые им альтерирующие эффекты на почечный ангиотензин II минимальны. Другими словами, почечные компенсаторные механизмы не затрагиваются. Это улучшает терапевтический индекс, позволяя назначать зилебесиран в повышенных дозах, не опасаясь развития неблагоприятных событий типа гипотензии, гиперкалиемии или острой почечной недостаточности, с которыми зачастую сталкиваются пациенты, принимающие стандартные гипотензивные препараты, блокирующие ренин-ангиотензин-альдостероновую систему (РААС), поскольку такие лекарства нацелены на нижележащие относительно ангиотензиногена компоненты сигнального пути РААС.
Опять же, зилебесиран позволит избавиться от избыточности в количестве используемых пациентами гипотензивных препаратов разных классов.
Следует, разумеется, тщательно изучить ряд важных вопросов, таких как не исключаемая потеря эффективности ввиду возможного появления антител к зилебесирану, его долгосрочная безопасность, более широкие преимущества в контексте сопутствующих заболеваний вроде сахарного диабета 2-го типа и хронической болезни почек.
Особняком стоит момент с возможностью быстрого отключения зилебесирана, если вдруг возникнет такая необходимость, к примеру, ввиду неожиданно сильного и устойчивого снижения артериального давления. Впрочем, создание малой интерферирующей РНК (миРНК), комплементарной миРНК зилебесирана особых трудностей не представляет.
Помимо грядущей опорной клинической программы фазы III, которая окончательно установит безопасность и эффективность зилебесирана среди широкой популяции пациентов, «Алнайлам фармасьютикалс» (Alnylam Pharmaceuticals) задумала проверить его способность снижать риск серьезных сердечно-сосудистых событий (MACE).
Предположительно, «Алнайлам» в начале коммерческого пути зилебесирана будет ориентировать его на лечение неконтролируемой гипертонии (она характеризуется скачкообразностью давления, недостаточным его снижением в ночное время, плохой приверженности пациентов терапии), которая устойчива к назначению трех или четырех гипотензивных препаратов разных классов. В дальнейшем, не исключено, будут подключены гипертоники с сердечной недостаточностью или диабетической нефропатией. В целом зилебесиран вполне способен стать новым стандартом лечения первичной (эссенциальной) гипертонии.
Коммерческий запуск зилебесирана на ключевых фармацевтических рынках планеты (включая США, Европу, Японию) ожидается приблизительно в 2030 году, причем с расширенным спектром терапевтических показаний, включающим в том числе снижение сердечно-сосудистой заболеваемости и смертности.
[/membership]
ЗИЛЕБЕСИРАН: КОЕ ЧТО ЕЩЕ
В свое время «Алнайлам фармасьютикалс» (Alnylam Pharmaceuticals) планировала разработку РНК-интерференционного препарата, в одной молекуле сочетающего две малые интерферирующие РНК (миРНК), чтобы одновременно осуществлять сайленсинг двух генов: ангиотензиногена (ANG) и ангиопоэтин-подобного белка 3 (ANGPTL3). Последний является валидированной мишенью в задаче снижения уровня атерогенных липидов.
Предполагаемый препарат, предназначенный для снижения риска серьезных сердечно-сосудистых событий (MACE) у высокорисковых пациентов, должен был снижать систолическое артериальное давление как минимум на 10 мм рт. ст. и уровень холестерина липопротеинов низкой плотности (ЛПНП) и триглицеридов хотя бы на 40%.
Параллельный сайленсинг реализовывался на рельсах технологической платформы GEMINI, а чрезвычайно редкое подкожное дозирование (один раз в год) — благодаря технологической платформе IKARIA.
Идея, высказанная еще в 2021 году, предполагала выбор препарата-кандидата в 2023 году. Однако, приоритеты, похоже, поменялись: ввиду бешеного спроса на препараты для лечения ожирения «Алнайлам» задумала заняться именно ими.
IONIS PHARMACEUTICALS И ПЕРЕДОВОЕ ЛЕЧЕНИЕ ГИПЕРТОНИИ
Вначале «Айонис фармасьютикалс» (Ionis Pharmaceuticals) пробовала разобраться с экспериментальным препаратом эвазарсен (evazarsen), который, по аналогии с зилебесираном (zilebesiran) авторства «Алнайлам фармасьютикалс» (Alnylam Pharmaceuticals), таргетирован против ангиотензиногена (AGT) в печени и предназначен для лечения резистентной гипертонии, не поддающейся адекватному контролю стандартными гипотензивными препаратами.
Затем, когда были собраны убедительные доказательства терапевтической состоятельности эвазарсена, «Айонис» переключилась на разработку его усовершенствованной версии в лице тонламарсена (tonlamarsen).
ЭВАЗАРСЕН: МЕХАНИЗМ ДЕЙСТВИЯ
Подкожно назначаемый эвазарсен (evazarsen, IONIS-AGT-LRx) представляет собой антисмысловой олигонуклеотид (ASO), ингибирующий синтез ангиотензиногена (AGT) в печени.
[membership level=»2,3″ show_noaccess=»true»]
Эвазарсен, будучи в точности комплементарным матричной РНК (мРНК), кодирующей AGT, связывается с ее нетранслируемым участком, тем самым приводя к ее же разрушению рибонуклеазой. В итоге предотвращается трансляция и останавливается синтез ангиотензиногена.
Для таргетной доставки эвазарсена в печень его ASO-последовательность ковалентно прилинкована к N-ацетилгалактозаминовому (GalNAc) лиганду, связывающему асиалогликопротеиновый рецептор 1 (ASGR1) на гепатоцитах.
В доклинических исследованиях эвазарсена на моделях грызунов с гипертонией или острой почечной недостаточностью было продемонстрировано падение уровня циркулирующего AGT на 90% с устойчивым снижением артериального давления [1].
Предполагалось, что коммерческая версия эвазарсена будет применяться подкожными инъекциями один или два раза в месяц.
[/membership]
ТОНЛАМАРСЕН: МЕХАНИЗМ ДЕЙСТВИЯ
«Айонис фармасьютикалс» (Ionis Pharmaceuticals), внимательно отслеживающая успехи конкурентов, поняла, что следует в срочном порядке оптимизировать режим дозирования эвазарсена (evazarsen), ведь прямой соперник зилебесиран (zilebesiran), которым занимается «Алнайлам фармасьютикалс» (Alnylam Pharmaceuticals), продемонстрировал, что эффективно снижает артериальное давление даже при очень редком дозировании подкожными инъекциями один раз в полгода.
[membership level=»2,3″ show_noaccess=»true»]
Для этого была подготовлена улучшенная версия эвазарсена — тонламарсен (tonlamarsen, ION904, AGT-2.5-LRx).
Антисмысловой олигонуклеотид (ASO) тонламарсен, во-первых, ингибирует синтез ангиотензиногена (AGT) в печени в более чем 10 раз сильнее, чем эвазарсен, и, во-вторых, может назначаться существенно реже: например, один раз в два или три месяца.
Не исключено, удастся разработать пероральную рецептуру тонламарсена.
[/membership]
ЭВАЗАРСЕН: КЛИНИЧЕСКИЕ РЕЗУЛЬТАТЫ
«Айонис фармасьютикалс» (Ionis Pharmaceuticals) завершила два клинических испытания NCT03714776 и NCT04083222 фазы II (рандомизированных, двойных слепых, плацебо-контролируемых, многоцентровых), которые оценили эффективность и безопасность эвазарсена (evazarsen) в лечении первичной (эссенциальной) артериальной гипертензии. Эвазарсен применялся соответственно монотерапевтически и в качестве дополнительного препарата к уже назначаемым гипотензивным лекарствам.
[membership level=»2,3″ show_noaccess=»true»]
Первое исследование охватило взрослых пациентов (n=25) с гипертонией (систолическое артериальное давление [САД] 140–165 мм рт. ст.), придерживающихся стабильной терапии двумя гипотензивными препаратами, один из которых — ингибитор ангиотензинпревращающего фермента (иАПФ) или блокатор рецептора ангиотензина II (БРА), а второй — бета-блокатор, блокатор кальциевых каналов или диуретик. Участники должны были прекратить прием всех гипотензивных лекарств.
Второе исследование пригласило пригласило взрослых пациентов (n=26) с неконтролируемой гипертонией (САД 140–170 мм рт. ст.), следующих стабильным курсом гипотензивной терапии из двух–трех препаратов разных классов, как то: в обязательном порядке иАПФ или БРА, плюс один–два дополнительных лекарства, таких как бета-блокатор, блокатор кальциевых каналов или диуретик (не относящийся к калийсберегающим). Участники должны были продолжать прием гипотензивных лекарств.
На протяжении 8 недель пациентам назначали 80-мг дозу эвазарсена или плацебо — подкожной инъекцией один раз в неделю.
Уровень сывороточного ангиотензиногена (AGT) в группах эвазарсена снизился на 54±25% и 67±14% в первом и втором исследованиях соответственно — против его роста на 3±18% и 13±23% в группах плацебо (p<0,001). Статистически значимое расхождение начало отмечаться на 8-й день, оставаясь таким вплоть до 92-го и 78-го дня.
Назначение эвазарсена привело к численно большему относительно плацебо (но статистически незначимому) снижению САД и диастолического артериального давления (ДАД). Так, усредненное изменение САД (мм рт. ст.) составило −8 (95% ДИ [здесь и далее]: −17, +2) и −12 (−21, −4), тогда как ДАД — −1 (−8, +5) и −6 (−11, −1).
В группах эвазарсена большая пропорция пациентов, чем в контрольных группах, продемонстрировала снижение САД и ДАД не менее чем на 5, 10 или 15 мм рт. ст., а также вышла к целевым показателям ниже 140 и 90 мм рт. ст. соответственно.
Второе исследование также показало отсутствие существенных различий между исходами в зависимости от того, принимали ли пациенты два (65% испытуемых) или три (35%) гипотензивных препарата.
Применение эвазарсена характеризовалось приемлемой переносимостью. Не зафиксировано гипотензивных событий, гиперкалиемии или почечных нарушений.
«Айонис» также запустила пару клинических испытаний фазы IIb (рандомизированных, двойных слепых, плацебо-контролируемых, многоцентровых), которые проверили еженедельное подкожное назначение эвазарсена в различных дозах и среди различных популяций пациентов.
Так, ASTRAAS (NCT04714320) изучило эвазарсен при неконтролируемой резистентной гипертонии среди пациентов (n=160), уже принимающих три и более гипотензивных препарата, а ASTRAAS-HF (NCT04836182) протестировало эвазарсен в лечении пациентов (n=72) с хронической сердечной недостаточностью со сниженной фракцией выброса (HFrEF).
Исследования завершены, однако их результаты не опубликованы.
[/membership]
ТОНЛАМАРСЕН: КЛИНИЧЕСКИЕ РЕЗУЛЬТАТЫ
«Айонис фармасьютикалс» (Ionis Pharmaceuticals) провела тонламарсен (tonlamarsen) через клиническое исследование NCT05314439 фазы II (рандомизированное, двойное слепое, плацебо-контролируемое, многоцентровое) среди взрослых пациентов (n=48) с неконтролируемой первичной (эссенциальной) артериальной гипертензией (систолическое артериальное давление [САД]) 130–170 мм рт. ст.), стабильно принимающих хотя бы одно гипотензивное лекарственное средство.
[membership level=»2,3″ show_noaccess=»true»]
Участникам назначали тонламарсен (30, 60 или 90 мг) или плацебо — подкожными инъекциями каждые 4 недели на протяжении 3 месяцев.
На 15-й неделе снижение уровня сывороточного ангиотензиногена (AGT) составило усредненных 79% и медианных 86% в 90-мг подгруппе тонламарсена — против его снижения на 6% и 5% в группе плацебо (p=0,001), тем самым механистически подтвердив состоятельность дозирования тонламарсена один раз в месяц [1].
Снижение САД как минимум на 10 мм рт. ст. было отмечено среди 42% пациентов, получавших тонламарсен, — против 30% в группе контроля.
Применение тонламарсена характеризовалось приемлемой переносимостью. Не зафиксировано ни целевых нежелательных явлений (НЯ), таких как гиперкалиемия, почечная дисфункция, гипотензия, ни внецелевых НЯ, таких как печеночная дисфункция, тромбоцитопения.
[/membership]
КОНВЕЙЕР
Сейчас конкуренции «Алнайлам фармасьютикалс» (Alnylam Pharmaceuticals) и ее зилебесирану (zilebesiran), считай, нет.
[membership level=»2,3″ show_noaccess=»true»]
С учетом того, насколько скромна «Айонис фармасьютикалс» (Ionis Pharmaceuticals) в объявлении клинических успехов, всё идет к тому, что программы эвазарсена (evazarsen) и тонламарсена (tonlamarsen) будут по итогам свернуты. Проблема состоит в том, что эти препараты-кандидаты, должным образом блокируя ангиотензиноген (AGT) в печени, требуют существенно более частого дозирования, чем зилебесиран, то есть не обладают конкурентным преимуществом.
Экспериментальный конвейер представлен считанными молекулами, таргетированными на AGT и изучаемыми в лечении гипертонии.
Так, раннестадийную клиническую проверку проходит РНК-интерференционный (RNAi) препарат-кандидат BW-00163 китайской «Арго байофарма» (Argo Biopharma). В начале 2024 года «Новартис» (Novartis) заключила с «Арго» многомиллиардное соглашение: не исключено, договоренности включают BW-00163 [1] [2].
Китайская «Сучжоу Райбо лайф сайенсиз» (Suzhou Ribo Life Science) занимается доклиническим RNAi-препаратом RBD9079 [3].
Китайские «Сучжоу Синеджин байо» (Suzhou Senegene Bio) и «Инновент байолоджикс» (Innovent Biologics) пробуют силы с доклиническим RNAi-препаратом SGB-3908 (IBI-3016) [4] [5].
На доклиническом этапе находятся STP136G и STP237G — RNAi-молекулы авторства «Сирнаомикс» (Sirnaomics) с офисами в США и Китае. Первое лекарственное соединение нацелено на AGT, второе — одновременно на AGT и аполипопротеин C3 (APOC3) [6].
«Модерна» (Moderna) в сотрудничестве с «Мерк и Ко» (Merck & Co.) разработала mRNA-4157 (V940) — персонализированную противораковую вакцину, которая производится отдельно под каждого пациента с четким прицелом на специфику его заболевания.
На данном этапе собраны весьма обнадеживающие клинические результаты лечения прошедшей хирургическую резекцию меланомы, превосходящие стандартную терапию.
Продемонстрированы примечательные исходы лечения неоперабельного рака головы и шеи. Продолжается клиническая проверка терапии рака легкого, рака почки, рака мочевого пузыря, немеланомного рака кожи.
Механистическое обоснование концепции персонализированных противораковых вакцин выглядит разумным и состоятельным. Мол, они запускают доселе невиданный мощный противоопухолевый иммунный ответ, причем высокоспецифичный, учитывающий особенности онкопатологии каждого конкретного пациента. Но пока шквал потенциально благотворных T-клеточных реакций не превратится в устойчивую и продолжительную клиническую ремиссию, будущего у противораковых вакцин попросту нет. И здесь обломали зубы десятки и сотни разработчиков. Но у «Модерна», кажется, получилось.
Если всё и дальше будет идти по плану, индивидуализированная неоантигенная мРНК-онковакцина mRNA-4157 (V940) получит ускоренное регуляторное одобрение в 2025 году.
Относительно несложный процесс выпуска и применения mRNA-4157 (V940) идет вразрез с комплексным и дорогостоящим производственным циклом и назначением терапии T-клетками с химерным антигенным рецептором (CAR), препараты на базе которой стали настоящим прорывом в лечении запущенных онкогематологических заболеваний.
Тем временем явных успехов добилась немецкая «Байонтек» (BioNTech). Персонализированная противораковая мРНК-вакцина аутоген цевумеран (autogene cevumeran), изучаемая совместно с «Дженентек» (Genentech) в составе «Рош» (Roche), весьма достойно показала себя в лечении рака поджелудочной железы.
Персонализированная иммунотерапия обеспечила уверенное продление жизни для трети пациентов с протоковой аденокарциномой поджелудочной железы.
МЕХАНИЗМ ДЕЙСТВИЯ
mRNA-4157 (V940) представляет собой персонализированную противораковую вакцину, которая, следуя концепции прецизионной медицины, должна резко улучшить эффективность лечения онкологических заболеваний.
Из опухолей и образцов крови пациента выделяют только ему свойственные неоантигены — антигены, закодированные мутантными генами, специфичными для раковых клеток в данных новообразованиях (последние развиваясь и прогрессируя, мутируют в обязательном порядке). При этом в состав онковакцины входят равно как эпитопы неоантигенов, которые были обнаружены в ходе ex vivo экспериментов над иммунными клетками пациента, так и эпитопы неоантигенов из всего экзома, которые, согласно прогностическому биоинформационному алгоритму, являются иммуногенными, то есть способными запустить в организме благотворные иммуностимулирующие реакции.
In silico собирается мРНК-последовательность, кодирующая одновременно до 34 эпитопов (неоантигенный конкатемер). Далее мРНК-молекулы инкапсулируются в фирменную оболочку из липидных наночастиц (LPN), которая наделяет готовый онковакцинный препарат толерантностью, минимизирует токсичность при многократном введении, помогает укрыться от надзора иммунной системы и защититься от ферментативного распада.
После внутримышечного введения персонализированной противораковой вакцины в организм антигенпрезентирующие клетки (APC) захватывают и транслируют мРНК-инструкции с дальнейшей экспрессией соответствующих эпитопов на своей поверхности. Итогом становится индуцирование специфических иммунных ответов со стороны цитотоксических T-клеток CD8+ и T-клеток памяти CD4+.
Доставка в организм сразу целого множества опухолеспецифических антигенов (TSA) должна резко повысить вероятность успешных клинических исходов, поскольку иммунная система начинает генерировать многовекторный T-клеточный ответ на неоантигенные пептиды, которые ей были презентированы. Другими словами, иммунная система проходит «обучение», по итогам которого усиливается ее способность распознавать и уничтожать опухолевые клетки.
В терминологии «Модерна» мРНК-вакцина mRNA-4157 (V940) относится к индивидуализированной неоантигенной терапии (Individualized Neoantigen Therapy, INT).
Разработка mRNA-4157 (V940) осуществляется совместно с «Мерк и Ко» (Merck & Co.), с которой «Модерна» в июне 2016 года вошла в стратегическое соглашение: первая выплатила второй 200 млн долларов авансом [1]. В мае 2018 года партнерство было расширено, что повлекло за собой финансовое вливание дополнительных 125 млн долларов [2]. В октябре 2022 года долгосрочный альянс укрепился еще сильнее: «Мерк и Ко» полностью поверила в успех mRNA-4157 (V940) [3].
КЛИНИЧЕСКАЯ ПРОВЕРКА
Продолжающееся клиническое исследование KEYNOTE-603 (NCT03313778) фазы I (нерандомизированное, открытое, многоцентровое) является основополагающим для дальнейшей разработки персонализированной противораковой вакцины mRNA-4157 (V940), поскольку изучает ее применимость различными схемами для лечения широкого ассортимента солидных опухолей.
Исследование охватывает такие диагнозы, как немелкоклеточный рак легкого (НМРЛ), мелкоклеточный рак легкого (МРЛ), меланома, уротелиальная карцинома (рак мочевого пузыря), плоскоклеточная карцинома головы и шеи (не индуцированная вирусом папилломы человека), протоковая аденокарциномы поджелудочной железы, а также любые злокачественные новообразования с высокочастотной микросателлитной нестабильностью (MSI-H) или дефектами в системе репарации ошибочно спаренных оснований ДНК (dMMR): например, колоректальный рак, аденокарцинома желудка и пищевода, рак эндометрия.
Параллельно осуществляется клиническое исследование KEYNOTE-942 (NCT03897881) фазы IIb (рандомизированное, открытое, многоцентровое, международное) среди пациентов с меланомой кожи (высокорисковой, на стадии IIIB–D или IV), которая метастазировала в лимфоузел и затем прошла полную хирургическую резекцию, но всё еще обладает высоким риском рецидива.
На момент рандомизации заболевание участников должно было быть в состоянии ремиссии, без локорегионарного рецидива, отдаленных метастазов, метастазирования в головной мозг.
Поставлена задача выяснить, оправдано ли добавление персонализированной мРНК-онковакцины mRNA-4157 (V940) к «Китруде» (Keytruda, пембролизумаб), блокатору PD-1 авторства «Мерк и Ко» (Merck & Co.), в целях продления безрецидивной выживаемости (RFS), если сравнивать с назначением только пембролизумаба (pembrolizumab). Предпосылки для этого очевидны: в адъювантных условиях (после резекции) опухоль отсутствует, пациент еще не проходил слишком много курсов химиотерапии, а его иммунная система относительно здорова.
Относительно недавно запущены следующие клинические испытания, оценивающие комбинацию mRNA-4157 (V940) и «Китруды» в задаче продления бессобытийной выживаемости после хирургической резекции, то есть удержания пациента в статусе ремиссии:
INTerpath-001 (V940-001, NCT05933577) фазы III. Меланома кожи (на стадии IIB–C, III или IV) после хирургической резекции, без признаков заболевания, с высоким риском рецидива, ранее не проходившая системную терапию. mRNA-4157 (V940) + пембролизумаб — сравнение с пембролизумабом. Конечные точки: RFS, DMFS, OS.
INTerpath-002 (V940-002, NCT06077760) фазы III. Немелкоклеточный рак легкого (НМРЛ) [на стадии II, IIIA или IIIB/N2] после хирургической резекции, без признаков заболевания, прошедший хотя бы одну линию адъювантной платиносодержащей химиотерапии. mRNA-4157 (V940) + пембролизумаб — сравнение с пембролизумабом. Конечные точки: DFS, OS, DMFS, LCSS.
V940-004 (NCT06307431) фазы II. Почечно-клеточная карцинома (рак почки) после хирургической резекции, без признаков заболевания, с промежуточно-высоким или высоким риском рецидива, ранее не проходившая какой-либо лечение. mRNA-4157 (V940) + пембролизумаб — сравнение с пембролизумабом. Конечные точки: DFS, OS, DMFS.
V940-005 (NCT06305767) фазы II. Мышечно-инвазивная уротелиальная карцинома (рак мочевого пузыря) после хирургической резекции, с высоким риском рецидива, ранее не проходившая системную терапию. mRNA-4157 (V940) + пембролизумаб — сравнение с пембролизумабом. Конечные точки: DFS, OS, DMFS.
INTerpath-007 (V940-007, NCT06295809) фазы II/III. Плоскоклеточная карцинома кожи (на стадии II–IV без отдаленных метастазов), местнораспространенная, операбельная, ранее не проходившая какую-либо терапию. mRNA-4157 (V940) + пембролизумаб до и после хирургической резекции — сравнение с хирургической резекцией ± пембролизумаб. Конечные точки: EFS, ORR, FFS rate, pCR rate, mPR rate, DFS, DSS, OS.
Сокращения: DFS, выживаемость без признаков заболевания; DMFS, выживаемость без отдаленного метастазирования; DSS, выживаемость с учетом заболевания; EFS, бессобытийная выживаемость; FFS, свобода от хирургического вмешательства; LCSS, выживаемость, специфическая при раке легкого; mPR, большой патоморфологический ответ; ORR, частота общего ответа; OS, общая выживаемость; pCR, полный патоморфологический ответ; RFS, безрецидивная выживаемость.
Geneos Therapeutics придумала, как вылечить распространенную гепатоцеллюлярную карциному.
ЛЕЧЕНИЕ МЕЛАНОМЫ
По прошествии наблюдений на протяжении медианных 2 лет (23–24 месяца) за пациентами с меланомой из KEYNOTE-942 (NCT03897881) фазы IIb, которые получили либо mRNA-4157 (V940) с «Китрудой» (Keytruda, пембролизумаб), либо только пембролизумаб (pembrolizumab), экспериментальное комбинированное иммунотерапевтическое лечение клинически значимым (но не статистически!) образом улучшило безрецидивную выживаемость (RFS), снизив риск рецидива или смертельного исхода на 44%, если сравнивать с монотерапией пембролизумабом: отношение риска (hazard ratio, HR) 0,56 (95% ДИ [здесь и далее]: 0,31–1,02; двусторонний p=0,053) [1] [2] [3].
[membership level=»2,3″ show_noaccess=»true»]
В статусе RFS на протяжении 12 месяцев оказались 83% и 77% пациентов, 18 месяцев — 79% и 62% [4].
Непротиворечивое продление RFS, обеспеченное персонализированной мРНК-онковакциной с блокатором PD-1, было отмечено вне зависимости от уровня опухолевой мутационной нагрузки (TMB), балла опухолевого воспаления (TIS) и статуса PD-L1. Соответствующие показатели HR таковы [5]:
высокая TMB (175 мутаций на экзом, или ≥ 10 мутаций на мегабазу) и невысокая TMB: 0,65 (0,28–1,49) и 0,59 (0,24–1,42);
высокий TIS и невысокий TIS (отсечение 4,56): 0,58 (0,21–1,59) и 0,53 (0,25–1,10);
опухоли, положительные (балл CPS ≥ 1) и отрицательные по PD-L1: 0,49 (0,23–1,04) и 0,16 (0,04–0,69).
По истечении 18 месяцев наблюдений с отдаленным метастазированием меланомы или смертельным исходом столкнулись 8% пациентов в группе комбинированного лечения и 24% — монотерапии «Китрудой». Частоты выживаемости без отдаленного метастазирования (DMFS) на протяжении 12 месяцев составили 93% и 89%, 18 месяцев — 92% и 77% [6] [7] [3].
Таким образом, добавление индивидуализированной противораковой вакцины mRNA-4157 (V940) к блокатору PD-1 пембролизумабу обеспечило клинически значимое улучшение показателя DMFS, снизив риск развития отдаленных метастазов или смерти на 65% в сравнении с лечением только «Китрудой» (HR 0,35 [0,15–0,83]; односторонний p=0,0063).
Установлено, что если до лечения не обнаруживалась циркулирующая опухолевая ДНК (ctDNA), которая отражает наличие минимально остаточной болезни (MRD) после опухолевой резекции и является биомаркером безрецидивной выживаемости при высокорисковой меланоме, прошедшей хирургическую операцию, то и ответ на терапию был лучше [8].
Так, среди пациентов с исходным негативным ctDNA-статусом показатели RFS и DMFS получились лучше при сравнении комбинированного лечения с монотерапией: риск рецидива или смерти и риск отдаленного метастазирования или смерти снизились на соответствующих 77% (HR 0,23 [0,10–0,53]) и 95% (HR 0,05 [0,01–0,38]). Впрочем, и при позитивном ctDNA-статусе были отмечены тенденции к улучшению RFS и DMFS, обеспеченные сочетанной терапией. Однако небольшая пациентская выборка не позволила сделать окончательные выводы, что эффективность терапевтической персонализированной противораковой вакцины mRNA-4157 (V940) в целом не зависит от изначального статуса ctDNA.
Индивидуализированная неоантигенная терапия mRNA-4157 (V940) работала лучше стандартного лечения вне зависимости от статуса BRAF: мутантный или дикого типа.
После медианных 3 лет наблюдений выяснилось, что лечение меланомы при помощи mRNA-4157 (V940) с пембролизумабом, клинически значимым образом улучшило показатели RFS и DMFS, снизив риск рецидива или смерти на 49% (HR 0,51 [0,29–0,91]; односторонний номинальный p=0,0095) и снизив риск развития отдаленных метастазов или смерти на 62% (HR 0,38 [0,17–0,86]; односторонний номинальный p=0,0077), если сравнивать с назначением только пембролизумаба [9].
[/membership]
СУТЬ
Длительное применение блокаторов PD-1 в адъювантном лечении резецированной высокорисковой меланомы в целях предупреждения ее возвращения — стандартная и эффективная клиническая практика [1] [2] [3], хотя и сопровождающаяся высоким риском хронической токсичности [4]. Однако терапевтическая персонализированная противораковая вакцина mRNA-4157 (V940) уже готова, кажется, обновить эту парадигму.
[membership level=»2,3″ show_noaccess=»true»]
Во всяком случае получены надежные доказательства улучшения RFS и DMFS в сравнении с нынешними терапевтическими подходами.
Так, наблюдения в течение 3,5 лет за пациентами, прошедшими годичный курс пембролизумаба (pembrolizumab), установили, что в статусах RFS и DMFS оставались 60% и 65% человек, а после 5 лет наблюдений — 55% и 61% [5] [6]. В случае годичной терапии ниволумабом (nivolumab) 4-летние показатели RFS и DMFS оказались справедливыми для 52% и 59% пациентов [7].
То есть в долгосрочной перспективе приблизительно половина пациентов сталкиваются с возвращением меланомы (или смертью), что вынуждает переходить к следующим линиям терапии. Таким образом, перед mRNA-4157 (V940) стояла задача как можно более продолжительного по времени сдерживания рецидива (RFS), который может усугубиться метастазированием (DMFS). И мРНК-онковакцина с таким упреждением справилась, хотя, впрочем, пока без подтвержденного статистически значимого расхождения с монотерапией пембролизумабом. Скорее всего, на это повлияла скромная по охвату выборка испытуемых.
Тем временем пембролизумаб засвидетельствовал оправданность своего одновременно назначения в неоадъювантном и адъювантном периодах, то есть равно как до резекции, так и после нее [8]. Сочетание ипилимумаба (ipilimumab) с ниволумабом продемонстрировало собственную пригодность в неоадъюватном периоде при определенных условиях [9] [10]. Гипотеза состоит в том, что еще не удаленная опухоль имеет достаточную массу, чтобы вызвать адекватную активацию и экспансию системного противоопухолевого T-клеточного ответа — такого же по силе, как в случае иммунотерапии рецидивирующей меланомы [11].
Открытым и важным остается вопрос с продлением общей выживаемости (OS): пока нет надежных данных, продлевают ли жизнь блокаторы PD-1, притом что конечные точки RFS и DMFS являются лишь суррогатными для OS, и делать какие-то определяющие заключения на их основе можно лишь с натяжкой [12].
В любом случае «Модерна» надо постараться с развитием mRNA-4157 (V940): в постковидную эпоху больше нет сверхдоходов от продажи вакцин против SARS-CoV-2.
С точки зрения бизнеса «Модерна» поступила чрезвычайно разумно, что потратила длительное время на проведение клинического испытания фазы IIb, прежде чем в конце июля 2023 года запустила решающую фазу III [13], поскольку было слишком много неудачных клинических исследований противораковых вакцин. Не всем нравится подобный промежуточный подход к разработке, но консервативная стратегия оправдана.
Чрезвычайная ситуация, связанная с пандемией коронавирусной инфекции COVID-19, официально прекращена.
[/membership]
ЛЕЧЕНИЕ РАКА ГОЛОВЫ И ШЕИ
Промежуточный анализ клинических исходов KEYNOTE-603 (NCT03313778) фазы I в когорте лечения неоперабельной (метастатической или рецидивирующей) плоскоклеточной карциномы головы и шеи (HNSCC) [ротовой полости, ротоглотки, гортаноглотки, гортани], не индуцированной вирусом папилломы человека (HPV−), ранее не проходившей терапию какими-либо ингибиторами иммунных контрольных точек (ИИКТ), установил следующие результаты [1].
[membership level=»2,3″ show_noaccess=»true»]
На начало октября 2020 года, комбинация из индивидуализированной мРНК-онковакцины mRNA-4157 (V940) и PD-1-блокатора «Китруды» (Keytruda, пембролизумаб) обеспечила частоту общего ответа (ORR) на уровне 50% (n=5/10): полный ответ (CR) зафиксирован у 20% (n=2/10) пациентов, частичный ответ (PR) — у 30% (n=3/10). Стабилизация заболевания (SD) отмечена у 40% (n=4/10). С прогрессированием (PD) столкнулся один человек. Таким образом, частота контроля заболевания (DCR), как суммы CR, PR и SD, составила 90% (n=9/10).
Медиана длительности ответа (DoR) еще не созрела. Медиана выживаемости без прогрессирования (PFS) — 9,8 месяца.
Что примечательно, 4 из 5 респондентов ответили на лечение после двух доз пембролизумаба (pembrolizumab)— еще до назначения mRNA-4157 (V940). После того как они получили вакцину, ответы углубились: к примеру, два пациента с частичной ремиссией перешли к полной. Еще один человек, заболевание которого прогрессировало на фоне «Китруды», после вливания вакцины наконец-то засвидетельствовал частичный ответ.
Для сравнения: монотерапия пембролизумабом рецидивирующей или метастатической плоскоклеточной карциномы головы и шеи выводит ORR и медиану PFS к 14,6% и 2,0 месяца. Более того, экспериментальное вакцинное противораковое лечение превзошло стандартную первоочередную терапию, предполагающую назначение «Китруды» вместе с химиопрепаратами и выдающую ORR 36% и медиану PFS 4,9 месяца [2] [3] [4].
Любопытные выводы сделаны сообразно анализу предиктивных биомаркеров. Отмечена тенденция к благоприятному клиническому ответу у пациентов с опухолями, характеризующимися повышенным уровнем воспаления, о котором свидетельствуют баллы GEP (профиль генной экспрессии) и CYT (цитолитическая активность). При этом, однако, корреляции с опухолевой мутационной нагрузкой (TMB) не выявлено.
Балл GEP, во-первых, отражает уровень РНК-экспрессии 18 воспалительных генов, имеющих отношение к антигенной презентации, экспрессии хемокинов, цитолитической активности и адаптивной иммунорезистентности (включая PD-L1), и, во-вторых, указывает на уровень T-клеточного воспаления в опухолевом микроокружении (TME). Балл CYT рассчитывается на основе транскрипционных уровней двух ключевых цитолитических эффекторов — гранзима A и перфорина.
Дальнейший анализ на начало мая 2023 года с медианой наблюдений в течение 9,0 (2,7–45,5) месяца и охвативший больше пациентов, установил следующие исходы: ORR 27% (11–50), включая CR 9% и PR 18%, DCR 64% (41–83), расчетные медианы PFS 3,5 (2,7–9,0) месяца и общей выживаемости (OS) 25,0 (9,0–NE) месяца [5].
[/membership]
СУТЬ
Пациенты с плоскоклеточной карциномой головы и шеи (HNSCC), отрицательной по вирусу папилломы человека (HPV−), характеризуются плохим прогнозом: пятилетняя частота выживаемости не превышает 50% [1].
[membership level=»2,3″ show_noaccess=»true»]
Считается, что ограниченный рост стойких клинических ответов на терапевтическую блокаду PD-(L)1 обусловлен снижением эффекторной цитолитической активности и клональным разнообразием [2] [3] [4] [5].
Поскольку индивидуализированная мРНК-онковакцина mRNA-4157 (V940), которая кодирует до 34 опухолевых неоантигенов, вызывает специфическую активацию противоопухолевых T-клеток, что было подтверждено успешным лечением меланомы, имело смысл опробовать ее назначение в связке с PD-1-блокатором пембролизумабом (pembrolizumab).
Иммуноонкологический коктейль продемонстрировал признаки активации иммунных реакций, которые обозначили себя среди ответивших на лечение в том числе снижением уровня циркулирующей опухолевой ДНК (ctDNA). «Более теплые» опухоли, такие как в случае HNSCC (HPV−), располагают более благоприятным опухолевым микроокружением (TME) для генерации T-клеточных ответов, индуцированных сочетанием mRNA-4157 (V940) c пембролизумабом. Кроме того, добавление неоантигенов, принесенных в составе персонализированной противораковой вакцины, снижает планку уровня опухолевой мутационной нагрузки (TMB), необходимого для должного терапевтического ответа на пембролизумаб.
Предварительные выводы таковы: при лечении HNSCC (HPV−) добавление мРНК-вакцины к стандартному пембролизумабу продлит общую выживаемость как минимум вдвое, если сравнивать с использованием только последнего.
[/membership]
ПРОТИВОРАКОВЫЕ ВАКЦИНЫ: ХОРОШО, ДА МАЛО
Несмотря на многообещающий потенциал противораковых вакцин, это направление биотехнологий развивается чрезвычайно медленно. Существует ряд препятствующих проблем: гетерогенность опухолевых антигенов и их мутационный разброс, отсутствие унифицированного сигнального пути распознавания антигенов с последующей активацией иммунного ответа, множество способов ухода раковых клеток от иммунологического надзора, выбор нужного иммуностимулирующего адъюванта, оптимальный путь доставки вакцины в организм.
Тем не менее на клиническом конвейере есть немало перспективных противораковых вакцин, включая персонализированные.
Ну а пока что в мире одобрены только две персонализированные противораковые вакцины: «Онкофаг» (Oncophage, витеспен) и «Провендж» (Provenge, сипьюлейсел-T).
Есть еще тройка противораковых препаратов на основе онколитических вирусов — «Онкорин» (Oncorine), «Имлайджик» (Imlygic, талимоген лагерпарепвек) и «Делитакт» (Delytact, тесерпатурев), применяемых в лечении соответственно карциномы головы и шеи, меланомы и злокачественной глиомы, — но к онковакцинам их можно отнести с натяжкой. Впрочем, механизм действия (хотя их несколько) этих лекарственных средств весьма близок: иммунной системе презентируются опухолевые антигены, накапливающиеся ввиду целенаправленного разрушения раковых клеток, индуцированного онколитическими вирусами.
Особняком стоит противотуберкулезная вакцина БЦЖ (бацилла Кальметта — Герена), которая почти полвека с большим успехом применяется в иммунотерапии рака мочевого пузыря без инвазии в мышечный слой (NMBIC).
«Онкофаг», ранее известная как «Профаг» (Prophage), представляет собой комплекс, включающий белок теплового шока (HSP) gp96 и фрагментов пептидов, полученных из опухолевой ткани пациента. Аутологичный антигенный комплекс витеспен (vitespen, HSPPC-96) стимулирует резидентные дендритные клетки, которые активируют цитотоксические T-лимфоциты (CTL) и T-хелперы — ключевые компоненты каскада противоопухолевого иммунного ответа [1].
«Онкофаг» разработана «Антидженикс» (Antigenics), которая в январе 2011 года сменила название на «Адженус» (Agenus) [2].
«Онкофаг» получила регуляторное разрешение только в России, где была одобрена в апреле 2008 года [3] [4].
В ноябре 2009 года «Антидженикс» отозвала заявку на регистрацию «Онкофага» в Европейском союзе, после того как экспертный комитет при Европейском агентстве по лекарственным средствам (EMA) пришел к заключению, что онковакцина не в силах существенно продлить безрецидивную выживаемость. Кроме того, производитель не предоставил полной информации относительно состава «Онкофага» и его производственного процесса, а также механизма действия при почечно-клеточной карциноме и выбора нужной дозы [5].
Применение «Онкофага» снижает риск прогрессирования заболевания или смерти (PFS) на 41% (HR 0,59 [0,37–0,94]; p=0,026) и риск смерти (OS) на 46% (HR 0,54 [0,30–0,97]; p=0,036) [6] [7].
[/membership]
«ПРОВЕНДЖ»
Персонализированная противораковая вакцина «Провендж» (Provenge, сипулейцел-T) предназначена для лечения бессимптомного или минимально симптоматического метастатического кастрационно-резистентного рака предстательной железы.
[membership level=»2,3″ show_noaccess=»true»]
«Провендж» производится из собственных мононуклеарных клеток периферической крови (PBMC) пациента, включая антигенпрезентирующие клетки (APC), T- и B-лимфоциты, естественные киллеры (NK), которые затем активируются ex vivo рекомбинантным человеческим белком PAP-GM-CSF. Последний представляет собой композицию из простатической кислой фосфатазы (PAP) — антигена, высокоэкспрессирующего в опухолевых тканях при раке простаты, и гранулоцитарно-макрофагального колониестимулирующего фактора (GM-CSF) — активатора иммунных клеток. После внутривенного вливания сипулейцела-T (sipuleucel-T, APC-8015) запускаются процессы гуморального и T-клеточного иммунного противоопухолевого ответа [1] [2].
«Провендж» разработана «Дендрион» (Dendreon), которую в феврале 2015 года за 495 млн долларов купила канадская «Валеант фармасьютикалс интернешнл» (Valeant Pharmaceuticals International), уже в июле 2017-го продавшая ее за 820 млн долларов китайскому конгломерату Sanpower Group [3] [4] [5] [6]. Через год последняя перепродала «Дендрион» за 868 млн долларов китайской Nanjing Cenbest, крупному розничному ретейлеру, решившему заняться биотехнологическим бизнесом. Впрочем, сделка по сути номинальная, ведь Sanpower является крупнейшим акционером Nanjing Cenbest [7].
В апреле 2010 года «Провендж» была одобрена Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) [8].
В сентябре 2013 года «Провендж» получила маркетинговое разрешение Европейского агентства по лекарственным средствам (EMA), однако в мае 2015-го регистрация была прекращена по просьбе «Дендрион» ввиду коммерческих соображений [9].
Терапевтическая эффективность «Провендж» весьма скромная: персонализированная противораковая вакцина, снижая риск смерти на 22% (HR 0,78 [0,61–0,98]; p=0,032), продлевает жизнь на медианных 4,1 месяца относительно плацебо [10]. И это притом что за курс лечения из 3 доз необходимо выложить почти 200 тыс. долларов при отсутствии страхового медицинского покрытия [11].
Рак поджелудочной железы — онкологическое заболевание, очень плохо поддающееся лечению.
Хирургическую резекцию опухолей может пройти лишь одна пятая пациентов.
Даже после успешного устранения опухолей почти всегда наступает рецидив, быстро ведущий к смерти.
Персонализированная противораковая вакцина на три года отсрочила рецидив и смерть у трети пациентов.
Открытыми остаются вопросы с доступностью такого прорывного метода лечения и его стоимостью.
ЧТО ПРОИЗОШЛО
Немецкая «Байонтек» (BioNTech) разрабатывает терапевтическую персонализированную противораковую вакцину, предназначенную для лечения онкологических заболеваний.
На данном этапе мРНК-онковакцина аутоген цевумеран (autogene cevumeran) зарекомендовала себя в успешном лечении рака поджелудочной железы, прошедшего полную резекцию.
По прошествии трех лет после ее применения приблизительно треть пациентов остаются в статусе полной ремиссии.
Это невероятное достижение, учитывая высочайшую агрессивность протоковой аденокарциномы поджелудочной железы, которая неминуемо возвращается даже после успешной хирургической операции.
Безоговорочные оптимистичные выводы делать преждевременно. Но уже понятно, что наметились предпосылки для выхода из стагнации, которая тянется вот уже шесть десятков лет кряду: при абсолютно любых методах лечения рака поджелудочной железы исходы весьма неблагоприятны.
Параллельно аутоген цевумеран проверяется в терапии других онкологических заболеваний, включая меланому и колоректальный рак.
Усилия «Байонтек» поддерживаются «Дженентек» (Genentech) в составе «Рош» (Roche). В конце сентября 2016 года партнеры договорились о совместной разработке персонализированных РНК-вакцин против рака. Швейцарский фармацевтический гигант пообещал заплатить 310 млн долларов авансом и последующими выплатами по мере реализации задуманного. Расходы и доходы делятся пополам [1].
Тем временем схожую работу проворачивают «Модерна» (Moderna) и «Мерк и Ко» (Merck & Co.), экспериментальная мРНК-онковакцина mRNA-4157 которых весьма неплохо справилась с лечением меланомы и рака органов головы и шеи.
мРНК-технологии рисуют оптимистичное будущее в лечении онкологических заболеваний.
ПОЧЕМУ ЭТО ВАЖНО
В более чем 95% случаев рака поджелудочной железы речь идет о ее протоковой аденокарциноме. На момент постановки диагноза у большинства пациентов отмечаются отдаленные метастазы или местнораспространенное неоперабельное заболевание, и только 20% больных характеризуются локализованной операбельной опухолью [1].
Рак поджелудочной железы является седьмой причиной смерти от онкологических заболеваний во всём мире [2]. С учетом роста заболеваемости [3] и почти неизменившейся за последние 60 лет выживаемостью на скромном уровне 12% [4] считается, что к 2025 году рак поджелудочной железы станет причиной еще большего числа смертей [3] [5].
Хирургия — единственный метод излечения рака поджелудочной железы. Однако у приблизительно 90% пациентов наблюдается рецидив заболевания в период медианных 7–9 месяцев после полной резекции, а пятилетняя общая выживаемость (OS) оказывается справедливой всего лишь для 8–10% человек [6] [7].
Хотя адъювантная мультилекарственная химиотерапия, являющаяся стандартом послеоперационного лечения, приводит к отсрочке рецидива, он всё равно возникает где-то через 14 месяцев [8]. Пятилетняя OS не превышает 30% [9]. Лучевая, биологическая и таргетная терапия неэффективны [8].
ГИПОТЕЗА
Протоковая аденокарцинома поджелудочной железы практически полностью нечувствительна к ингибиторам иммунных контрольных точек, таким как блокаторы PD-L(1): частота ответа на лечение не превышает 5% [1] [2].
Частично это объясняется тем, что опухоль характеризуется низким уровнем мутаций, то есть образует мало неоантигенов [2]. Последние представляют собой уникальные белковые последовательности, появляющиеся в результате процесса мутации опухоли, и маркируют опухолевые клетки как чужеродные для T-клеток. Другими словами, малое количество неоантигенов делает рак поджелудочной железы слабо антигенным с небольшим количеством инфильтрирующих T-клеток, которые уничтожают опухолевые клетки.
Однако недавние наблюдения установили, что неоантигенов при протоковой аденокарциноме [3] [4] [5] всё же больше, чем ранее предполагалось [6].
Исследования долгожителей при раке поджелудочной железы [7] [8] засвидетельствовали, что неоантигены способны стимулировать T-клетки в протоковой аденокарциноме. Первичные опухоли, обогащенные иммуногенными неоантигенами, содержали приблизительно в 12 раз более высокую плотность активированных Т-клеток CD8+, что коррелирует с замедлением рецидива заболевания и более продолжительной выживаемостью пациентов.
Таким образом, поскольку в большинстве случаев протоковой аденокарциномы ее опухоли содержат неоантигены, способные стимулировать Т-клетки, выдвинута гипотеза о стратегии доставки в организм неоантигенов, индуцирующих неоантиген-специфические Т-клетки с целью улучшения исходов лечения, включая устранение микрометастазов и сдерживание рецидива.
Терапевтические мРНК-вакцины, подкрепленные адъювантами, оптимально подходят для проверки этой гипотезы [9].
Необходимая мРНК-последовательность, кодирующая множество неоантигенов, быстро изготавливается в виде персонализированных под каждого пациента вакцин [10], которые активируют антигенпрезентирующие клетки [11] [12] [13] [14]. Эффективная доставка мРНК-вакцины в организм отлажена в ходе разработанных клинических препаратов [15].
Geneos Therapeutics придумала, как вылечить распространенную гепатоцеллюлярную карциному.
ДОКАЗАТЕЛЬСТВО
Клиническое исследование NCT04161755 фазы I (нерандомизированное, открытое, многоцентровое) пригласило взрослых пациентов (n=16) с протоковой аденокарциномой поджелудочной железы (T1–3, N0–2, M0), прошедших полную хирургическую резекцию. Макроскопическая полнота (R0 и R1) последней подтверждалась гистологически.
Испытуемые получали следующее последовательное лечение:
«Тецентрик» (Tecentriq, атезолизумаб): блокатор PD-L1 авторства «Рош» (Roche) — по прошествии 6 недель после резекции, одна 1200-мг доза.
Аутоген цевумеран (autogene cevumeran, BNT122, RO7198457): индивидуализированная мРНК-онковакцина, подготовленная на фирменной биотехнологической платформе iNeST (individualized Neoantigen Specific Therapies), заключенная в наночастицы липоплекса и содержащая до 20 иммунодоминантных неоантигенов, высокоаффинных главным комплексам гистосовместимости класса I (MHC-I) и MHC класса II (MHC-II) [1] [2], — по прошествии 9 недель после резекции, еженедельно по 25 мкг (8 примирующих доз и 1 бустерная доза).
Химиотерапевтическая схема mFOLFIRINOX: модифицированная схема из четырех препаратов (фолиниевая кислота, фторурацил, иринотекан, оксалиплатин) — по прошествии 21 недели после резекции, 12 циклов.
Идея состояла, во-первых, в амплификации неоантиген-специфических Т-клеток, подавленных сигнализацией PD-1, во-вторых, в примировании наивных Т-клеток к вакцинным неоантигенам, в-третьих, синергичном эффекте системных химиопрепаратов.
По истечении медианных 18 месяцев наблюдений результаты получились следующими [3].
В когорте пациентов (n=19), среди которых оценивалась безопасность лечения, медианы общей выживаемости (OS) и безрецидивной выживаемости (RFS) достигнуты не были.
Что касается когорты пациентов (n=16), среди которых оценивались биомаркеры, то у 8 человек, ответивших на терапию аутогеном цевумераном (респондентов), медиана RFS достигнута не была, тогда как у 8 неответивших на лечение (нереспондентов) она вышла к 13,4 месяца: отношение риска (hazard ratio [HR]) 0,08 (95% ДИ [здесь и далее]: 0,01–0,40; p=0,003).
Другими словами, назначение терапевтической персонализированной противораковой мРНК-онковакцины на 92% снизило риск рецидива или смертельного исхода при раке поджелудочной железы.
Ландмарк-анализ, который был необходим для исключения систематической ошибки, связанной со временем до ответа, и который соотнёс RFS с ответом у пациентов, которые не столкнулись с рецидивом на протяжении получения всех 8 доз аутогена цевумерана, также установил отсутствие медианы RFS среди респондентов — против 11,0 месяца среди нереспондентов: HR 0,06 (0,008–0,4; p=0,008).
У респондентов уровень CA19-9 в сыворотке крови — наиболее широко используемый клинический биомаркер рака поджелудочной железы [4] — был стабильно ниже, если сравнивать с нереспондентами.
Для исключения вероятности того, что респонденты были просто пациентами с лучшим прогнозом, было подтверждено, что ответ на атезолизумаб (atezolizumab), позитивность по лимфатическим узлам, позитивность по краям среза, размер первичной опухоли, количество доз химиотерапии и плотность внутриопухолевых T-клеток CD8+ никак не коррелировали с ответом на мРНК-вакцину.
Респонденты и нереспонденты характеризовались сопоставимой иммунологической пригодностью, поскольку они выдали эквивалентные гуморальные и клеточные ответы на другую мРНК-вакцину (против коронавируса SARS-CoV-2), которая вводилась одновременно с аутогеном цевумераном.
У респондентов и нереспондентов были эквивалентные частоты всех основных циркулирующий клеток врожденного и адаптивного иммунитета, а также сходные соматические и зародышевые генеративные характеристики.
Таким образом, T-клеточный ответ, усиленный аутогеном цевумераном, не только коррелировал с отсроченным рецидивом рака поджелудочной железы, но и не был связан с различиями в отборе пациентов, частотах внутриопухолевых T-клеток или T-клеток периферической крови, общей пригодности для лечения.
ОБНОВЛЕНИЕ
По прошествии медианных 3 лет (2,3–3,8) наблюдений респонденты продолжали демонстрировать продление RFS (ее медиана достигнута не была), тогда как этот показатель для нереспондентов остановился на медианных 13,4 месяца: HR 0,14 (0,03–0,60; p=0,007) [1] [2]. То есть риск рецидива или смерти снизился на 86%.
Два респондента столкнулись с рецидивом: у них было выявлено меньше совокупных T-клеток, индуцированных вакциной, по сравнению с респондентами без рецидива.
По итогам 75% (n=6/8) респондентов продолжали оставаться в статусе ремиссии, тогда как 88% (n=7/8) нереспондентов столкнулись с рецидивом.
Согласно секвенированию вариабельных регионов бета-цепей T-клеточных рецепторов (TCRVβ), осуществленному до вакцинации и после нее, аутоген цевумеран (autogene cevumeran) индуцировал 79 различных неоантиген-специфических клонов T-клеток CD8+ с предполагаемой продолжительностью жизни медианных 5,5 года (1,3–70,2): медианных 8 (2–28) клонов на пациента. Поскольку 98% таких клонов отсутствовали до вакцинации, предполагается, что терапевтическая персонализированная противораковая вакцина индуцировала их de novo.
По прошествии 3 лет большинство (85%) новых T-клеточных клонов продолжали персистировать в организме респондентов, что, скорее всего, и определило продление их безрецидивной выживаемости в сравнении с нереспондентами.
ЧТО ДАЛЬШЕ
«Байонтек» продолжает изучать стратегии повышения уровня и глубины терапевтического ответа на применение терапевтической персонализированной противораковой вакцины аутоген цемуверан (autogene cevumeran). Это включает дальнейшую оптимизацию ее потенциала и расширение пространства идентификации неоантигенов для включения в состав мРНК-онковакцины генетических аберраций за пределами однонуклеотидных полиморфизмов (вариаций, инсерций, делеций): например, слияний.
Пока не закрыт вопрос с тем, вносит ли вакциноиндуцированное клональное разнообразие T-клеток вклад в устойчивый контроль над заболеванием [1] [2]. Тем не менее отмечено, что опухоли у ответивших на вакцину были более клональными, представляя собой, возможно, опухоли в иммуноопосредованном развитии — как это наблюдалось у долгожителей с раком поджелудочной железы [3].
Предположительно, более клональная первичная опухоль отражает способность иммунной системы распознать опухоль, то есть ответить на вакцину. Опять же, факты того, что качество неоантигенов [3] [4] [5] — маркер опухолей с наиболее иммунодоминантными неоантигенами — коррелирует с иммуногенными вакцинными неоантигенами, дополнительно поддерживают концепцию о необходимости оптимального выбора неоантигенов.
Проверка терапевтической персонализированной противораковой вакцины аутоген цемуверан была осуществлена в адъювантных (послеоперационных) условиях потому, что исторически вакцины против каких-либо инфекционных патогенов демонстрировали наибольшую эффективность в профилактических задачах, а не терапевтических. Связано это, вероятно, с тем, что эффективность вакцин требует оптимально функционирующей иммунной системы хозяина.
Активная опухоль, патологическая сигнализация которой нарушает должную работу иммунной системы, и недостаточность информации о гетерогенности неоантигенов между опухолями могут препятствовать ответам на противораковую вакцинацию. Вот почему «Байонтек» целенаправленно придерживается подхода клинической проверки аутогена цемуверана среди пациентов с минимально остаточной болезнью, чтобы вакцина смогла по-максимуму отсрочить рецидив или смерть.
В случае пациентов с протоковой аденокарциномой поджелудочной железы, прошедших ее полную резекцию, находящихся в статусе без каких-либо признаков остаточного заболевания и еще не получавших первоочередное системное лечение, проводится клиническое исследование IMCODE003 (NCT05968326) фазы II, в котором назначение тройной комбинации из аутогена цемуверана, «Тецентрика» и mFOLFIRINOX сравнивается с применением стандарта в лице mFOLFIRINOX.
Параллельно осуществляется клиническое исследование NCT04486378 фазы II среди пациентов с высокорисковым колоректальным раком (стадия II–III), которые прошли полную резекцию и у которых обнаруживается циркулирующая опухолевая ДНК (ctDNA), то есть они могут столкнуться с рецидивом в течение двух–трех лет после хирургического вмешательства. Назначение аутогена цемуверана сравнивается со стандартной в данном случае тактикой «наблюдения и ожидания».
Особняком стоит клиническое исследование IMCODE001 (NCT03815058) фазы II, изучающее первоочередное лечение метастатической (рецидивирующей или de novo на стадии IV) или неоперабельной местнораспространенной (стадия IIIC–D) меланомы сочетанием аутогена цемуверана с «Китрудой» (Keytruda, пембролизумаб), блокатором PD-1 авторства «Мерк и Ко» (Merck & Co.). Группа контроля получает только стандартный пембролизумаб (pembrolizumab).
Ведущей экспериментальной онкологической вакциной является GT-30, изучаемая в иммуноонкологическом лечении распространенного рака печени (гепатоцеллюлярной карциномы).
Терапия второй линии, осуществленная после первоочередного назначения стандартных «Нексавара» (Nexavar, сорафениб) или «Ленвимы» (Lenvima, ленватиниб), вывела приличную пропорцию пациентов с неоперабельным или метастатическим раком печени к статусу ремиссии, тем самым открыв возможность для проведения потенциально излечивающего хирургического вмешательства наряду с облучением.
С позиции продления жизни пациентов клиническая результативность онковакцины GT-30 превзошла все одобренные фармакологические подходы к лечению распространенного рака печени.
В целом ситуация медленно, но верно улучшается. Так, если четыре десятка лет назад пятилетняя относительная выживаемость при раке печени в США составляла крошечных 3%, сейчас этот показатель подрос до 22% [1]. Но этого решительно недостаточно: данный показатель идет вслед за относительной выживаемостью в ничтожных 13% при раке поджелудочной железы [2].
Как бы то ни было, несмотря на впечатляющий прогресс медицинской науки, говорить о победе над раком печени еще очень и очень рано.
В реальности нет смысла лечить неоперабельный рак печени препаратами.
ПОЧЕМУ ЭТО ВАЖНО
Гепатоцеллюлярная карцинома, будучи наиболее распространенной формой первичного рака печени [1], является одной из основных причин смерти от онкологических заболеваний во всём мире [2]. Ежегодно ставится свыше 860 тыс. диагнозов рака печени и умирает более чем 760 тыс. человек [3] [4]. В большинстве случаев гепатоцеллюлярная карцинома возникает на фоне цирроза печени и связана с определенными факторами риска, такими как вирусный гепатит B или C, чрезмерное употребление алкоголя, а также неалкогольная жировая болезнь печени (НАЖБП), ассоциированная с метаболическим синдромом и сахарным диабетом [5] [6] [7] [8]. Рак печени в 60% случаев выявляется не на ранней стадии, не подходящей для проведения оперативного вмешательства, которое в теории способно его вылечить [9] [10] [11] [12]. Существующие лекарственные препараты против распространенной гепатоцеллюлярной карциномы характеризуются весьма скромной терапевтической эффективностью, по сути не предоставляющей пациентам какого-либо ощутимого шанса остаться в живых [13] [14] [15] [16].
ПОДХОД
«Джинеос терапьютикс» (Geneos Therapeutics), будучи дочерним биотехнологическим стартапом «Иноувио фармасьютикалс» (Inovio Pharmaceuticals), официально появилась в поле зрения инвесторов в феврале 2019 года. На сегодня «Джинеос» привлекла стороннее финансирование в размере 44,5 млн долларов [1] [2].
Если материнская «Иноувио» погружена в «популярные» заболевания вроде дисплазий, вызванных вирусом папилломы человека (ВПЧ), или коронавирусной инфекции COVID-19, то «Джинеос» сделала ставку на те патологии, к которым весьма трудно подобраться ввиду явных сложностей с лечением. Сейчас стартап сосредоточен на лечении двух онкологических состояний: распространенной гепатоцеллюлярной карциномы и впервые диагностированной глиобластомы с неметилированным промотором MGMT.
«Джинеос» лицензировала у «Иноувио» фирменные технологии создания ДНК-вакцин и их доставки в организм.
В рамках процесса SynCon генерируется ДНК-последовательность, несущая отобранные антигены и оптимизированная в целях улучшения стабильности мРНК, загрузки рибосом и экспрессии антигенных белков. Вакцинный препарат, представляющий собой плазмиды, в которые встроена готовая ДНК-последовательность, внутрикожно или внутримышечно вводится в организм.
Для того чтобы плазмиды проникли в клетки используется устройство «Селлектра» (Cellectra), организующее электропорацию, когда ряд кратковременных электрических импульсов заставляет клеточные мембраны открываться. Через несколько часов или дней клетки начинают синтезировать антигены — их презентирование иммунной системе запускает продуцирование антител и T-киллерных клеток, «заряжая» организм возможностью борьбы с опухолевыми клетками.
«Джинеос» объединила эти технологии в лице персонализированной иммунотерапевтической платформы GT-EPIC, на рельсах которой организовано создание специфичной для каждого пациента вакцинной ДНК-плазмиды, кодирующей неоантигены (ненормальные мутации или вариации генома, продуцируемые раковыми клетками), полученные из РНК опухолевых биоптатов.
Онковакцина затем сочетается с другой ДНК-плазмидой, кодирующей цитокин интерлейкин 12 (IL-12), в данном случае выступающий адъювантом. Комбинированная молекула внутрикожно доставляется в организм электропорационным устройством «Селлектра». Иммуноонкологический терапевтический коктейль активирует неоантигеноспецифические T-хелперы CD4+ и киллерные T-клетки CD8+, целенаправленно уничтожающие опухолевые клетки.
Персонализированная иммунотерапия обеспечила уверенное продление жизни для трети пациентов с протоковой аденокарциномой поджелудочной железы.
СИЛЬНЫЕ СТОРОНЫ
«Джинеос» уверена, что платформа GT-EPIC располагает рядом преимуществ перед решениями конкурентов, занимающихся персонализированным противораковым лечением, таким как неоантигенные вакцины и клеточная терапия.
Во-первых, плазмиды стимулируют ответы со стороны T-клеток CD4+ и CD8+, что характерно для вакцин на основе вирусных векторов, но уже сложнее для вакцин на базе белков или пептидов. Связано это с тем, что и плазмиды, и вирусные векторы вызывают экспрессию антигена внутри клеток: для генерации устойчивого и сильного T-клеточного CD8+ ответа антигенам требуется быть презентированными на молекулах класса I главного комплекса гистосовместимости (MHC-I), то есть процессинг и презентирование должно происходить внутриклеточно.
Во-вторых, плазмиды способны нести относительно высокую полезную нагрузку: их емкость позволяет кодировать существенно большее число различных антигенов в сравнении с вирусными векторами. В продолжающихся клинических испытаниях «Джинеос» использует до 40 неоантигенов для каждого пациента, но, согласно доклиническим исследованиям, без особых проблем можно закодировать гораздо больше таковых (даже 80). Концепция состоит в том, чтобы демонстрировать иммунной системе пациента как можно большее число неоантигенов-мишеней — она сама разберется, какие из них будут лучше стимулировать иммунный ответ, по итогам отразившийся должной клинической эффективностью.
В отрасли ведутся дебаты, могут ли неоантигены с неоптимальным противоопухолевым эффектом блокировать более продуктивные неоантигенные ответы посредством иммунодоминирования. «Джинеос», опирающаяся на клинические данные, придерживается позиции, что пациенты, получившее большее количество неоантигенов, отвечают на лечение лучше, чем получившее меньшее их число.
Если сравнивать ДНК-вакцины с мРНК-вакцинами, последние всё же оптимальнее с позиций доставки полезной нагрузки: им необходимо лишь пересечь плазматическую мембрану, дабы запустить производство белковых антигенов, тогда как первые должны дополнительно миновать ядерную оболочку, что снижает итоговую эффективность. Тем не менее преимущество ДНК-вакцин состоит в их большей стабильности (они могут годами храниться при температуре 2–8 ℃), то есть они практичнее с точки зрения дистрибуции в развивающихся странах и сельской местности, не располагающих должной холодовой инфраструктурой хранения готовых препаратов.
В-третьих, важным преимуществом платформы GT-EPIC является относительно низкая себестоимость продукции и более короткие сроки изготовления плазмид, если сравнивать с вакцинами на основе аденовирусных векторов или мРНК либо CAR-T-клеточной терапией. Ценовой аспект позволит по-настоящему демократизировать персонализированное лечение рака.
Опять же, высокая скорость создания готовых плазмид, укладывающаяся в 6–8 недель (впоследствии процесс ужмется до 3–4 недель), важна для пациентов с раком на поздних стадиях: такие больные, ввиду стремительного прогрессирования онкологического заболевания, не в силах ждать 4–6 месяцев, когда будет готов индивидуализированный лекарственный препарат. Оперативность подготовки плазмид также открывает возможность для лечения онкопатологии на ранних стадиях, не прибегая к хирургическому вмешательству.
НАЧАЛО
GT-30, ведущая экспериментальная противораковая вакцинная программа «Джинеос», ориентирована на лечение распространенной гепатоцеллюлярной карциномы посредством вышеописанной иммунотерапии, сочетанной с «Китрудой» (Keytruda, пембролизумаб), блокатором PD-1 авторства «Мерк и Ко» (Merck & Co.).
Рак печени был выбран по той причине, что в его «холодных» опухолях отсутствуют инфильтрирующие лимфоциты, то есть опухолевые клетки решительно недостаточно отвечают на назначение только ингибиторов PD-(L)1.
Применение GT-30 превращает опухоли в «горячие» иммуногенные путем примирования (запуска) ответа T-клеток на периферии, вдали от опухоли. Терапия преодолевает определенные ключевые механизмы резистентности, задействованные при раке печени, фактически «загоняя» T-клетки в опухолевые клетки.
В ноябре 2022 года «Джинеос» продемонстрировала, что применение GT-30 обеспечило частоту контроля над заболеванием (DCR) у 54% (n=13/24) пациентов, причем 13% (n=3/24) человек достигли полного ответа (CR), то есть вышли к ремиссии [1].
Для сравнения: CR при лечении рака печени только «Китрудой» не превышает 4%.
Согласно анализу репертуара T-клеточных рецепторов (TCR) в периферической крови и опухолевой ткани, осуществленному до и после иммуноонкологической вакцинации, у всех пациентов выявлены новые или размножившиеся клоны T-клеток, преимуществено с активированным фенотипом CD8+. К 9-й неделе лечения они проникали в микроокружение опухоли (TME), что, скорее всего, способствовало ее регрессии.
СЕЙЧАС
Клиническое исследование GT-30 (NCT04251117) фазы Ib/IIa (нерандомизированное, открытое, многоцентровое) охватило взрослых пациентов (n=36) с гепатоцеллюлярной карциномой, которые ранее получили первоочередное лечение тирозинкиназными ингибиторами «Нексаваром» (Nexavar, сорафениб) или «Ленвимой» (Lenvima, ленватиниб), в ответ на которое либо заболевание прогрессировало, либо развилась непереносимость.
Неоперабельная гепатоцеллюлярная карцинома испытуемых должна была находиться, согласно Барселонской системе стадирования рака печени (BCLC), либо на стадии C (распространенная стадия), либо на стадии B (промежуточная стадия), не подходящей для проведения локорегионарной терапии или рефрактерной к ней.
На момент анализа данных, который состоялся по прошествии наблюдений в течение медианных 21,5 месяца, 34 из 36 пациентов прошли хотя бы одно повторное сканирование, то есть для них имелась возможность оценить ответ на лечение в соответствии с критериями RECIST 1.1.
Два пациента прекратили терапию по причине несвязанных с ней тяжелых нежелательных явлений (НЯ), но были включены в полный анализ данных в качестве нереспондентов.
Частота общего ответа (ORR) составила 31% (n=11/36). К полному ответу (CR), то есть к статусу ремиссии рака печени, вышли 8% (n=3/36) испытуемых [1] [2] [3].
Дополнительно четвертому больному, заболевание которого изначально было неоперабельным, удалось полностью избавиться от всех опухолевых поражений (первичных в печени и метастазных в легких) — после пятой дозы терапевтической персонализированной противораковой вакцины, что открыло для него возможность пройти потенциально излечивающую хирургическую резекцию вкупе с облучением.
Частичный ответ (PR) был получен у 22% (n=8/36) человек, стабилизация заболевания (SD) была зафиксирована у 25% (n=9/36) испытуемых, с прогрессированием рака печени столкнулись 39% (n=14/36) участников.
Таким образом, частота контроля заболевания (DCR), как сумма CR, PR и SD, составила 56% (n=20/36).
Подтверждена оправданность использования ультрачувствительного анализа третьего поколения, оценивающего уровень циркулирующей опухолевой ДНК (ctDNA) и позволяющего относительно надежно установить текущий статус заболевания (прогрессирование или регрессирование).
Анализ ctDNA характеризуется высокой прогностической значимостью: снижение уровня ctDNA при раке печени обычно предшествует улучшению результатов на МРТ, тем более что очаги гепатоцеллюлярной карциномы, если судить по МРТ, зачастую не разрешаются полностью, несмотря на отсутствие клинических признаков остаточного заболевания.
Медиана длительности ответа (DOR) не достигнута, медиана выживаемости без прогрессирования (PFS) составила 4,2 месяца, медиана общей выживаемости (OS) получилась равной 19,9 месяца.
Экспериментальное лечение характеризовалось приемлемым профилем безопасности, в целом не отличающимся от применения только пембролизумаба. Серьезных НЯ после назначения противораковой вакцины зарегистрировано не было. Наиболее частыми НЯ были реакции по месту инъекций, которые носили преходящий характер легкой степени тяжести.
мРНК-технологии рисуют оптимистичное будущее в лечении онкологических заболеваний.
СУТЬ
Наблюдаемый в настоящее время научный прогресс в области разработки и клинической противораковых мРНК-вакцин характеризуется тем, что такие препараты применяются почти исключительно в адъювантном режиме: для предотвращения рецидива после хирургического удаления опухоли. И потому складывается мнение, будто терапевтические персонализированные онковакцины эффективны лишь после резекции, но никак не до ее проведения, то есть они якобы не работают при лечении распространенных, неоперабельных или метастатических опухолей. Но это ошибочно, если судить по обнадеживающим результатам применения ДНК-вакцины GT-30 авторства «Джинеос», которая наделила полными ответами пациентов с поздними стадиями прогрессирующего рака печени, который хирургическое вмешательство не проходил.
ИТОГИ
Лечение распространенной гепатоцеллюлярной карциномы при помощи GT-30, терапевтической персонализированной противораковой вакцины, нашло ответ у трети пациентов (ORR 31%), и это весьма приличный показатель.
Согласно ряду клинических испытаний моноприменения блокаторов PD-(L)1, таких как «Китруда» (Keytruda, пембролизумаб), «Опдиво» (Opdivo, ниволумаб), «Имфинзи» (Imfinzi, дурвалумаб) и «Тевимбра» (Tevimbra, тислелизумаб), изученных в первоочередном и второлинейном лечении распространенного рака печени, выдаваемая ими ORR укладывалась в пределы 12–18% [1] [2] [3] [4] [5] [6] [7] [8].
Более того, GT-30 обеспечила полный ответ в 8% случаев и медиану общей выживаемости на уровне 19,9 месяца, тогда как указанные ингибиторы иммунных контрольных точек (ИИКТ) вывели эти показатели к более скромным диапазонам в 2–4% и 13,2–16,6 месяца. Однако онковакцина не смогла существенным образом сдержать прогрессирование заболевания.
Моноприменение блокаторов PD-(L)1 практически не в силах выдать какой-либо полный ответ при гепатоцеллюлярной карциноме ввиду иммунной резистентности, обусловленной спецификой опухолевого микроокружения, и низкой опухолевой мутационной нагрузки (TMB) [9].
Зато персонализированная иммунотерапия, благодаря своей способности индуцировать опухолеспецифические T-клеточные ответы, сенсибилизирует опухоли к назначению ИИКТ, которые, «снимая тормоза» с иммунной системы, начинают работать должным образом [10] [11] [12] [13] [14] [15].
В целом состоятельность подхода, организованного «Джинеос», неоднократно подтверждалась разработчиками других терапевтических персонализированных противораковых вакцин: как в доклинических исследованиях [16] [17] [18] [19], так и в клинических испытаниях при меланоме, глиобластоме, раке поджелудочной железы, немелкоклеточном раке легкого, рак мочевого пузыря [20] [21] [22] [23] [24] [25] [26]. Однако именно «Джинеос» одной из первых уверенно доказала, что метод эффективно работает в случае гораздо менее чувствительных к иммунотерапии типах опухолей, таких как гепатоцеллюлярная карцинома.
Монотерапия моноклональными антителами против PD-(L)1, как известно, обращает вспять дисфункцию T-клеток в существующих неоантиген-специфических T-клеточных клонах, но не может индуцировать новые неоантиген-специфические T-клеточные клоны [27] [28].
Согласно иммунологическому анализу, добавление онковакцины GT-30 устранило это досадное ограничение, обеспечив как индукцию новых Т-клеточных ответов на кодируемые ею неоантигены, так и расширение репертуара T-клеточных рецепторов (TCR) в периферической крови и опухоли. Согласно анализу секвенирования отдельных клеток, основной вклад в рост T-клеточного пула был сделан со стороны эффекторных T-клеток памяти CD8+.
«Джинеос» не устает подчеркивать оправданность включения в терапевтическую онковакцину как можно большего числа различных целевых неоантигенов, не обходя стороной драйверные (driver) и пассажирские (passenger), основные (truncal) и дополнительные (branch), общие (shared) и частные (private) мутации, а также неоантигенные эпитопы с более широким диапазоном прогнозируемой аффинности связывания с молекулами I класса человеческого лейкоцитарного антигена (HLA). Максимально развернутый репертуар неоантигенов, доставляемых онковакциной, приводит к задействованию более широкого набора иммунных реакций, тем самым улучшая шансы на успешное лечение.
Попутно высказана гипотеза, что сопутствующие факторы, такие как микроокружение опухоли и иммунная приспособленность, могут играть определяющую роль в характере клинического ответа [29].
Ограничением онковакцины GT-30 является возможное раннее приобретение резистентности к ней вследствие неоантигенной потери и гетерогенности опухоли. Решением может стать сбор образцов опухолевой ДНК из нескольких очагов в сочетании с быстрым синтезом новых версий персонализированной вакцины в целях возвращения контроля над очагами, не реагирующими на терапию.
ПЕРСПЕКТИВЫ
Методы лечения неоперабельного рака печени стремительно развиваются, и потому, очевидно, в обозримом будущем появятся такие, которые обеспечат улучшенные клинические исходы, чем онковакцина GT-30.
Так, например, уже одобрены иммунотерапевтические комбинации из атезолизумаба (atezolizumab) и бевацизумаба (bevacizumab), дурвалумаба (durvalumab) и тремелимумаба (tremelimumab), камрелизумаба (camrelizumab) и ривоцераниба (rivoceranib), на которые пациенты весьма неплохо отвечают [1] [2] [3].
Однако у персонализированного лечения гепатоцеллюлярной карциномы, предложенного «Джинеос», есть неоспоримое преимущество, связанное с высокой безопасностью. Конкурирующие терапевтические подходы, напротив, характеризуются повышенным риском развития тяжелых или серьезных иммуноопосредованных нежелательных явлений, то есть для их применения подходят далеко не все пациенты. Вот почему онковакцина GT-30 непременно займет должное место в арсенале лечения рака печени.
БУДУЩЕЕ
Как и любое новое направление медицины, терапевтические персонализированные противораковые вакцины проходят длинный путь разработки и развития, сопровождающийся своими падениями и взлетами.
Эволюция в этой области напоминает историю нынешней иммунотерапии онкологических заболеваний, которая, без оглядки на обилие неудач в ходе экспериментов, в итоге всё же подарила надежду пациентам с солидными и гематонкологическими опухолями. Так, на протяжении почти 20 лет многочисленные клинические испытания моноклональных антител не демонстрировали воспроизводимой эффективности, прежде чем в 1997 году на сцену не вышел ритуксимаб (rituximab) [1]. Долгие годы блокаторы PD-(L)1 не показывали ощутимой клинической результативности — пока в 2008 году не были опубликованы первые результаты применения ниволумаба (nivolumab) [2]. В течение многих лет у CAR-T-клеток не получалось встать на рельсы успешной терапевтической модальности [3].
Онковакцины, индивидуализированные с учетом опухолевой специфики конкретного пациента, засвидетельствовали равно как собственное механистическое обоснование, так и убедительные клинические результаты. Они вплотную подошли к тому этапу становления, когда можно смело утверждать, что вскоре они примкнут к методам стандартной противораковой терапии.
Особенно преуспели «Модерна» (Moderna) и «Байонтек» (BioNTech), мРНК-онковакцины которых успешно прошли клиническую проверку лечения меланомы и рака поджелудочной железы соответственно.
Важнейшей темой для обсуждения остается выбор сроков проведения иммунотерапевтической вакцинации. Большинство персонализированных онковакцин исследуются на поздних стадиях заболевания и после применения ингибиторов иммунных контрольных точек (ИИКТ), однако, есть мнение, что они могут принести гораздо больше пользы, если будут подключаться на более ранних этапах терапии: например, в неоадъювантном периоде (до хирургического вмешательств), когда пациент еще не столкнулся с феноменом иммуноредактирования, истощением Т-клеток, дальнейшим ослаблением иммунной системы [4].