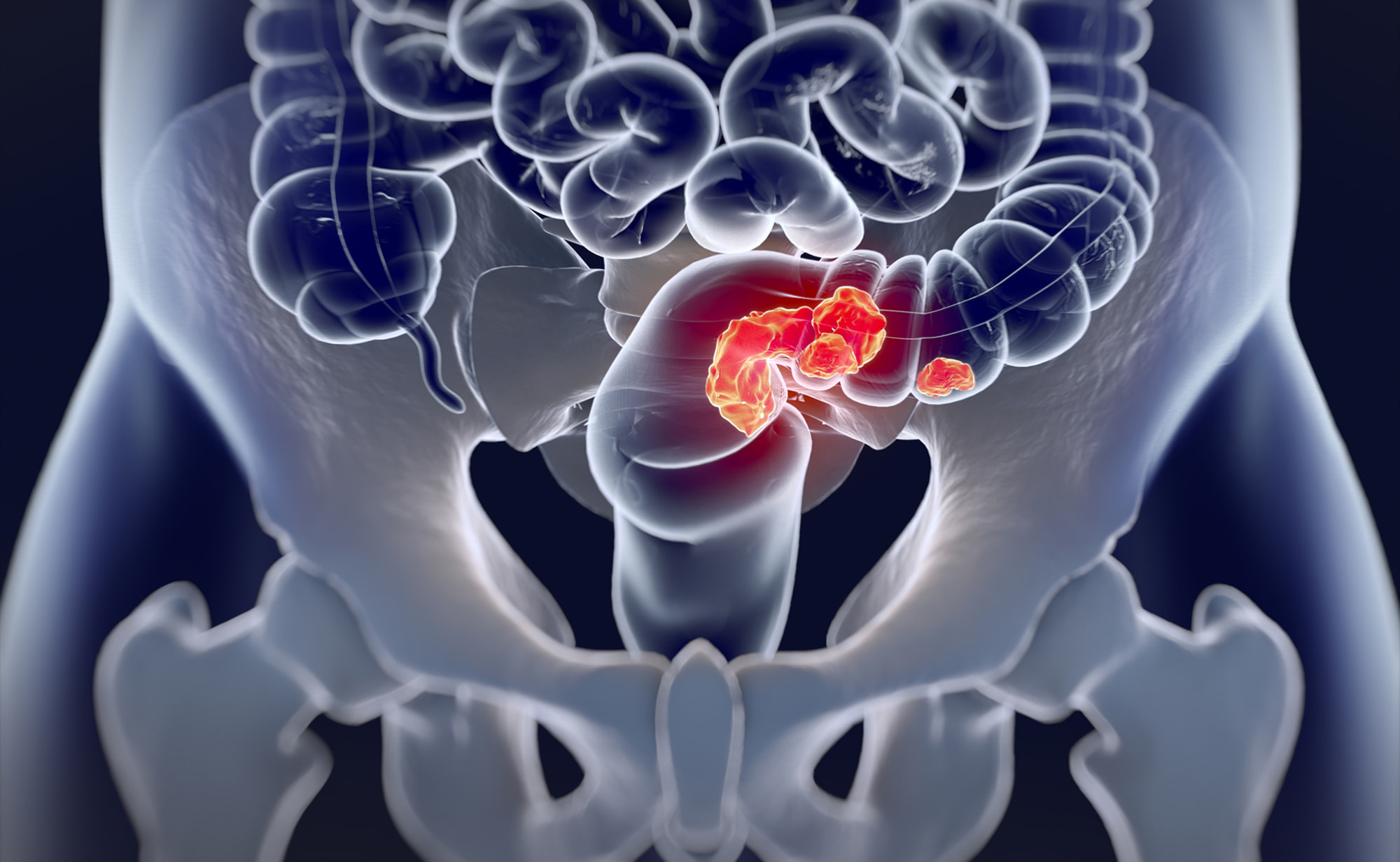
«Джемперли» (Jemperli, достарлимаб) обеспечил прежде невиданную ремиссию в ходе лечения рака прямой кишки.
ОСНОВНЫЕ ФАКТЫ
Шестимесячная монотерапия достарлимабом (dostarlimab) местнораспространенного рака прямой кишки с высокочастотной микросателлитной нестабильностью (MSI-H) или дефектами в системе репарации ошибочно спаренных оснований ДНК (dMMR) привела к тому, что после ее завершения больше половины пациентов, для которых были собраны клинические данные, вышли к стойкой ремиссии, которая продолжалась два года.
Никому из ответивших на экспериментальное лечение испытуемых не потребовалось последующее стандартное лечение.
«Джемперли» (Jemperli, достарлимаб), блокатор PD-1, предлагаемый «ГлаксоСмитКляйн» (GlaxoSmithKline), одобрен в лечении рецидивирующих или распространенных солидных опухолей с dMMR/MSI-H, включая рак эндометрия, идущий по отдельному терапевтическому показанию.
Иммунотерапевтический препарат достарлимаб продлит жизнь при распространенном или рецидивирующем раке эндометрия.
ПРЯМАЯ РЕЧЬ
«Достарлимаб, обеспечивший полную ремиссию на протяжении двух лет, должен стать совершенно новым подходом к лечению местнораспространенного рака прямой кишки с MSI-H/dMMR, не требующего стандартного вмешательства с его изнурительными для пациента последствиями».
Андреа Сёрсек (Andrea Cercek), заведующая отделением колоректального рака и содиректор Центра колоректального и желудочно-кишечного рака с ранним началом Мемориального онкологического центра им. Слоуна — Кеттеринга (MSK, Нью-Йорк, США).
«Собранные клинические данные приближают нас к пониманию потенциала достарлимаба в условиях местнораспространенного рака прямой кишки с MSI-H/dMMR. Беспрецедентная 100-процентная частота клинически полных ответов подтверждает грядущее изменение парадигмы лечения этой формы онкологии».
Хешам Абдулла (Hesham Abdullah), старший вице-президент и руководитель глобальных онкологических исследований и разработок «ГлаксоСмитКляйн» (GlaxoSmithKline).
СУТЬ ВОПРОСА
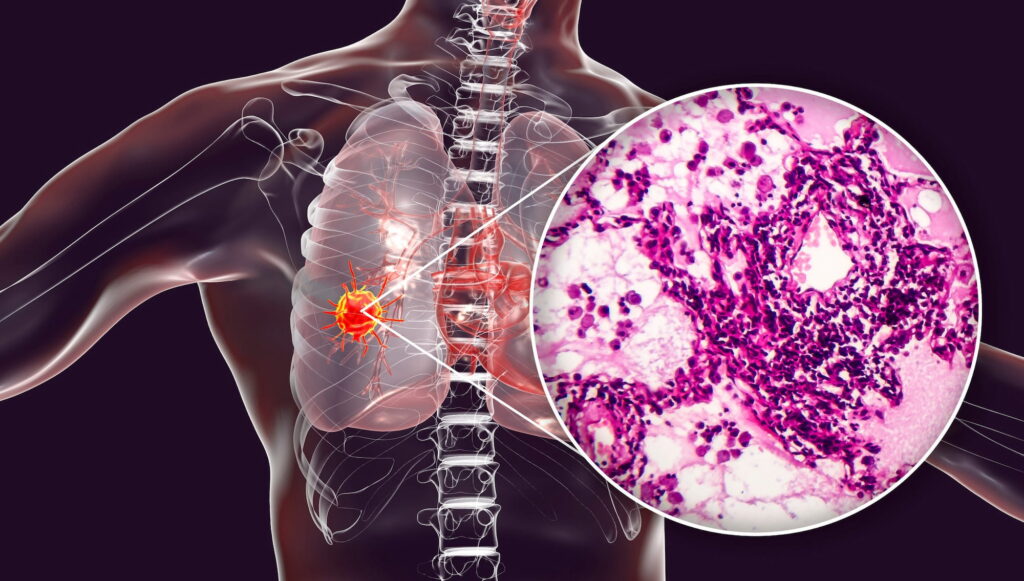
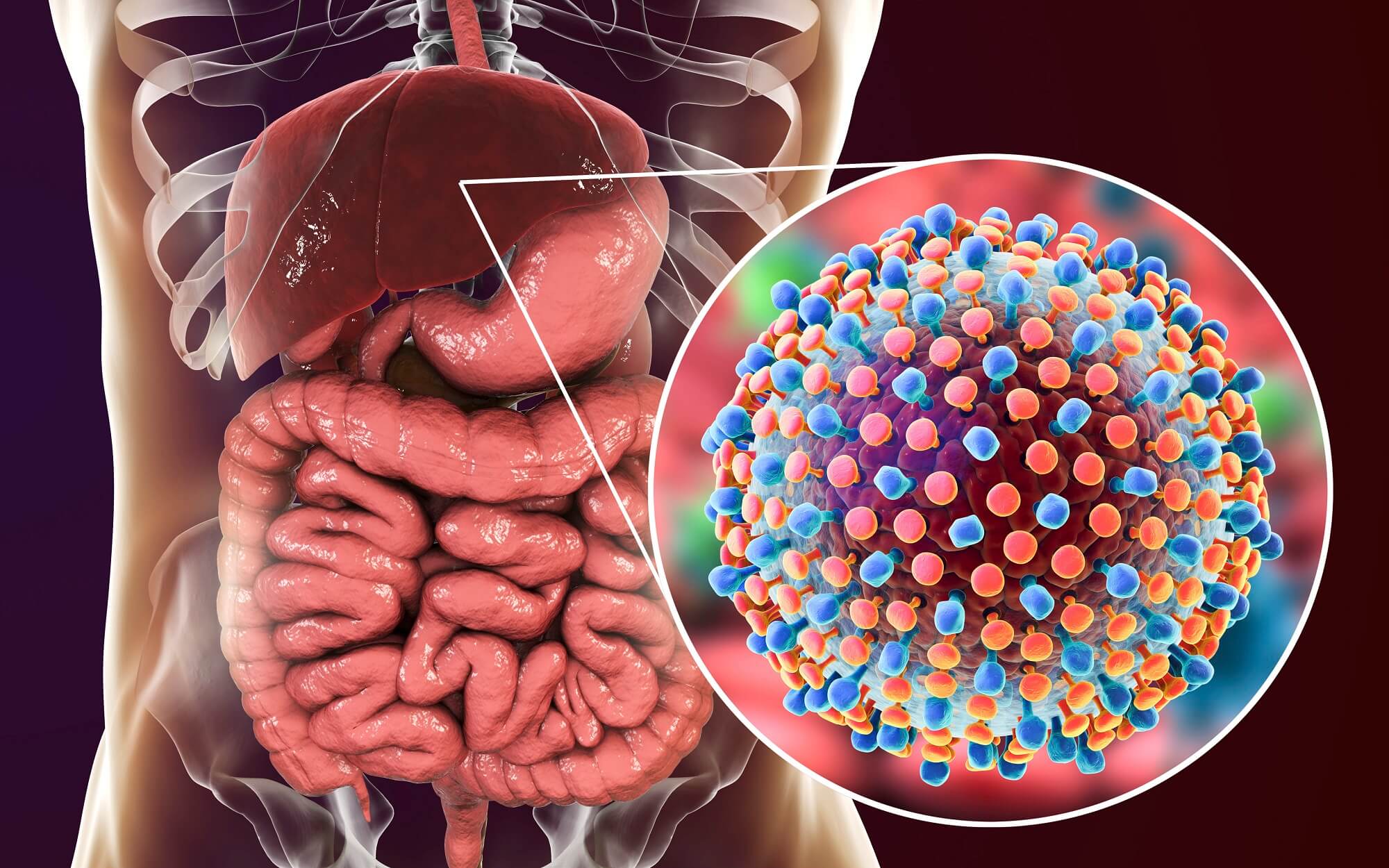
Рак прямой кишки — это форма рака, которая возникает в конечном отделе толстого кишечника и которая обычно относится к группе онкологических заболеваний, называемых колоректальным раком.
Колоректальный рак занимает третье место в мире по частоте диагностирования [1]. Приблизительно в 5–10% случаев рака прямой кишки отмечаются опухолевые аномалии dMMR/MSI-H, влияющие на корректность восстановления ДНК при ее копировании в клетке [2]. Такие аномалии являются биомаркером, который предсказывает успешный ответ на терапевтическую PD-(L)1-блокаду иммунных контрольных точек [3] [4]. Подобные опухоли чаще всего встречаются при раке эндометрия, колоректальном раке и других желудочно-кишечных онкологических заболеваниях, но могут обнаруживаться и при иных солидных опухолях [5] [6] [7] [8].
ПОЧЕМУ ЭТО ВАЖНО
Нынешний стандарт лечения местнораспространенного рака прямой кишки с MSI-H обращается к первоочередному назначению химиотерапии с облучением, затем проводится опухолевая резекция вместе с участками кишечника и/или окружающих тканей [1]. Это приводит к положительным результатам для большинства пациентов, однако почти одна треть больных в конечном итоге умирает ввиду отдаленного метастазирования [2]. Кроме того, оперативное вмешательство и химиорадиотерапия могут отражаться долгосрочными неблагоприятными последствиями, оказывающими серьезное негативное влияние на качество жизни по причине нарушения функции кишечника и мочеиспускания, половой дисфункции, вторичных раковых заболеваний, бесплодия [1].
КЛИНИЧЕСКИЕ ПОДРОБНОСТИ
Клиническое исследование NCT04165772 фазы II (нерандомизированное, открытое, многоцентровое) привлекло взрослых пациентов (n=16) с прежде нелеченной местнораспространенной ректальной аденокарциномой на стадии II/III с MSI-H/dMMR.
Протокол испытания предполагал, что участники получат неоадъювантный (до хирургического вмешательства) достарлимаб (каждые 3 недели на протяжении 6 месяцев), после чего, в случае остаточного заболевания, будет проведена стандартная лучевая терапия с одновременным назначением химиотерапевтического капецитабина, а затем, в случае сохранения заболевания, — тотальное мезоректальное иссечение.
По прошествии медианных 12 месяцев (6–25) наблюдений за пациентами (n=12), которые прошли лечение достарлимабом, результаты получились следующими [1]:
клинически полный ответ (cCR) составил 100% (95% ДИ [здесь и далее]: 74–100);
опухоль не обнаруживалась никаким из доступных способов, включая МРТ-изображения, ПЭТ-изображения, эндоскопию, пальцевое ректальное исследование, биопсию;
прогрессирования или рецидива заболевания не зафиксировано;
никому из пациентов не потребовались последующие химиорадиотерапия или хирургическое вмешательство.
[su_spoiler class=»my-custom-spoiler» title=»Эволюция эндоскопического и радиографического ответа у пациентов с раком прямой кишки, получавших лечение достарлимабом (dostarlimab). Предупреждение: медицинские изображения могут показаться неприемлемыми.»]
[/su_spoiler]
Столь впечатляющие результаты заставили «ГлаксоСмитКляйн» расширить набор пациентов, чтобы убедиться в высокой эффективности достарлимаба, и она не прогадала.
Так, по прошествии медианных 17,9 месяца (0,3–50,5) наблюдений за испытуемыми (n=42), которые полностью прошли 6-месячный курс лечения при помощи «Джемперли», cCR был зарегистрирован для всех 100% человек [2] [3].
Кроме того, по истечении медианных 26,3 месяца (12,4–50,5) наблюдений, все 24 пациента, для которых был доступен анализ данных, оставались в статусе устойчивого cCR, продолжавшегося как минимум 12 месяцев.
В целом медианное время до фиксирования клинически полного ответа составило 6,22 месяца (6,18–6,45).
ЧТО ДАЛЬШЕ
Продолжается клиническое испытание AZUR-1 (NCT05723562) фазы II, призванное подтвердить успехи достарлимаба в монотерапии прежде нелеченного местнораспространенного рака прямой кишки на стадии II/III (T3–T4, N0 или Tx, N+) с MSI-H/dMMR. Собираются данные, касающиеся пропорции пациентов, сохраняющих статус полной ремиссии по прошествии 12, 24 и 36 месяцев после лечения.
Осуществляется также клиническое исследование AZUR-2 (NCT05855200) фазы III, сравнивающее достарлимаб с химиотерапией в первоочередном лечении колоректального рака на стадии II (T4N0) или стадии III (операбельного) с MSI-H/dMMR. Накапливаются данные по частоте бессобытийной выживаемости (EFS) на протяжении 5 лет после терапии.
ЭКСПЕРТНЫЕ КОММЕНТАРИИ
Победа индукционного неоадъювантного назначения достарлимаба над раком прямой кишки обнадеживает. Облучение и хирургическое вмешательство имеют необратимые последствия для фертильности, сексуального здоровья, функции кишечника и мочевого пузыря [1] [2] [3] [4] [5]. Существенны также последствия для качества жизни, что особенно актуально для молодых людей детородного возраста, поскольку среди них растет заболеваемость раком прямой кишки [6].
Следует, впрочем, понимать, что аденокарцинома прямой кишки только в 5–10% случаев характеризуется наличием MSI-H/dMMR. Такие опухоли весьма плохо отвечают на стандартные схемы химиотерапии, включая неоадъювантную [7] [8] [9].
Важный вопрос заключен в том, почему локализованные опухоли прямой кишки с MSI-H/dMMR реагируют на имммуноонкологическое лечение гораздо сильнее, чем метастатические колоректальные опухоли с MSI-H/dMMR [10].
Одно из объяснений заключается в иммуномодулирующем влиянии микробиома кишечника: некоторые виды бактерий усиливают противоопухолевый иммунный ответ, потенцированный неоадъювантным назначением ингибитора иммунных контрольных точек (ИИКТ) [11] [12] [13]. В ряде клинических испытаний ИИКТ в лечении немелкоклеточного рака легкого (НМРЛ), меланомы и почечно-клеточной карциномы было подтверждено существенное благотворное влияние определенных бактерий кишечного микробиома на противоопухолевый ответ [14] [15] [16].
Кроме того, различия в ответах могут быть связаны с характеристиками опухолевых клеток помимо чрезвычайно высокого мутационного бремени (ввиду нарушенной системы репарации), такими как клональность, анеуплоидия и класс мутации [17] [18] [19].
Если разбивать медианное время до регистрации клинически полного ответа, согласно МРТ прямой кишки, эндоскопии, биопсии, уровню циркулирующей опухолевой ДНК (ctDNA) или ПЭТ-КТ, то оно получилось соответственно равным 6,15 месяца (6,09–6,25), 6,18 месяца (3,62–6,22), 1,41 месяца (1,38–2,73), 1,38 месяца (1,38–2,76) и 2,76 месяца (2,73–6,18).
Получается, что наиболее прогностически ранним оказался высокочувствительный анализ на ctDNA: изначально она обнаруживалась у 97% пациентов, а уже через 6 недель лечения перестала выявляться у более половины участников.
Продолжение наблюдений за пациентами из NCT04165772, чтобы установить окончательную продолжительность ремиссии, позволит выяснить, избавляет ли неоадъюватное иммуноонкологическое лечение от необходимости в хирургической резекции в долгосрочной перспективе.
В будущем, возможно, неоадъюватная блокада PD-1 окажется востребованной в лечении других опухолей с MSI-H/dMMR, таких как локализованный рак поджелудочной железы, желудка, предстательной железы. Следует подтвердить гипотезу востребованности высокоактивной противораковой терапии в неоадъювантном контексте до химиорадиотерапии и хирургического вмешательства, то есть до воздействия других агентов, которые могут нацелиться на клетки с резистентным фенотипом.
Фармакологические усилия борьбы с болезнью Паркинсона путем избавления от нейротоксичного альфа-синуклеина выглядят не особо впечатляющими с точки зрения терапевтической эффективности — тем не менее игроки фармотрасли не сдаются.
СУТЬ ВОПРОСА
Болезнью Паркинсона, вторым после болезни Альцгеймера самым распространенным нейродегенеративным возрастным заболеванием, страдают свыше 10 млн человек в мире. К 2040 году число больных, как ожидается, достигнет 17 млн человек, приобретя форму пандемии [1] . Несмотря на многочисленные исследовательские усилия, болезнь Паркинсона по-прежнему остается неизлечимой [2].
Отличительными патологическими особенностями болезни Паркинсона являются дегенерация дофаминергических нейронов и отложения телец Леви, состоящих в основном из нерастворимых агрегатов, образованных неправильно свернутым альфа-синуклеином (синуклеопатия) — небольшим белком, состоящий из 140 аминокислот.
В последние годы гипотеза агрегации альфа-синуклеина привлекает большое внимание. Она постулирует, что в патологических условиях альфа-синуклеин подвергается мисфолдингу (неправильному сворачиванию), что приводит к его конформационным изменениям, вызывающим аномальную агрегацию. В ходе этого процесса образуются различные виды агрегатов альфа-синуклеина, включая олигомеры, протофибриллы и фибриллы, все из которых цитотоксичны для нейронов и способствуют их гибели. Особенно губительны промежуточные олигомеры и протофибриллы, причем растворимые протофибриллы обладают большей нейротоксичностью, чем нерастворимые зрелые фибриллы [3] [4].
Токсичные промежуточные продукты ответственны за запуск каскада событий, включая дисфункцию митохондрий, нейровоспаление, деформацию нейронов и ферроптоз, которые тесно связаны с патогенезом болезни Паркинсона. Они нарушают синаптическую передачу, функции органелл и целостность цитоскелета, структуру мембран и гематоэнцефалического барьера (ГЭБ) [5] [6].
Если говорить о митохондриях, которые являются основным источником реактивных форм кислорода (ROS) в клетках, то они особенно уязвимы для агрегатов альфа-синуклеина. Последние задействуют механизмы связывания с комплексами митохондриальной дыхательной цепи, взаимодействия с митохондриальными порами переходной проницаемости, вмешательства в импорт митохондриальных белков, нарушения контроля качества митохондрий [7].
Агрегация альфа-синуклеина нарушает работу аутофагического лизосомального пути (ALP) и убиквитин-протеасомной системы (UPS) — двух основных механизмов деградации неправильно свернутых и агрегированных белков, — что отражается изменениями внутриклеточного транспорта и снижением скорости клиренса поврежденных белков [8] [9].
В физиологических условиях микроглия и астроциты функционируют как соответственно ключевые иммунные и опорные клетки центральной нервной системы (ЦНС). Однако в присутствии патологического альфа-синуклеина пролиферация микроглии стимулируется такими механизмами, как активация толл-подобных рецепторов (TLR) и сигнальных путей p38/ATF2 и ядерного фактора κB (NF-κB). Всё это результирует выработкой различных факторов воспаления и ROS, способствуя дисфункции и гибели дофаминергических нейронов [10]. Альфа-синуклеиновые агрегаты активируют микроглию, которая дифференцируется в провоспалительный фенотип (фенотип M1) и вырабатывает провоспалительные факторы, хемокины и нейротоксические факторы, которые побуждают астроциты трансформироваться в нейротоксический фенотип (фенотип A1) [11], еще больше усиливающий активацию микроглии [12].
Альфа-синуклеиновые агрегаты распространяются по организму в первую очередь «прионоподобным» образом. Они также могут распространяться через туннелирующие нанотрубки (нанотуннели между клетками) [13], экзосомы [14] и посредством иных механизмов, что приводит к повсеместному отложению этих патологических белков по всему головному мозгу, особенно в неокортексе, гиппокампе, стриатуме, таламусе и мозжечке [6]. Аномальная агрегация альфа-синуклеина не ограничивается мозгом: она наблюдается в спинном мозге и периферической нервной системе, включая симпатические ганглии, блуждающий нерв, сердечные нервы и желудочно-кишечную систему [15].
Согласно гипотезе «кишечник — мозг», при болезни Паркинсона патологические процессы зарождаются в кишечнике и впоследствии распространяться на головной мозг [16]. Альфа-синуклеин вызывает изменения в составе микробиоты кишечника и кишечное воспаление, что приводит к дисфункции иммунной системы кишечника, энтеральной нервной системы и кишечного барьера. Активация глиальных клеток кишечника, повышенная кишечная проницаемость и окислительный стресс повышают уровень экспрессии альфа-синуклеина в кишечнике, что отражается мисфолдингом и аномальной агрегацией этого белка. Хроническое периферическое воспаление нарушает целостность ГЭБ, способствуя переносу альфа-синуклеина из кишечника в ЦНС [17] [18].
GLP1R-агонисты вроде ликсисенатида, эксенатида или семаглутида сдерживают прогрессирование моторных нарушений при болезни Паркинсона.
ПРАСИНЕЗУМАБ: PROTHENA / ROCHE
Ирландская «Проутина» (Prothena) совместно с «Рош» (Roche) доводит до ума гуманизированное моноклональное антитело прасинезумаб (prasinezumab, RG7935, PRX002, NEOD002) против агрегированного альфа-синуклеина. Но результаты пока не впечатляют.
Прасинезумаб, оригинатором которого является, «Неотоуп байосайенсиз» (Neotope Biosciences), связывается с человеческим агрегированным альфа-синуклеином с высоким сродством и авидностью. На мышиных моделях болезни Паркинсона и деменции с тельцами Леви прасинезумаб снижал уровень C-концевой усеченной формы альфа-синуклеина, которая считается нейротоксичной, а также сдерживал распространение альфа-синуклеина от клетки к клетке [1] [2] [3].
Клиническое исследование PASADENA (NCT03100149) фазы II, организованное среди пациентов (n=316) с недавно диагностированной болезнью Паркинсона (длительность заболевания не дольше 2 лет) и не придерживающихся заместительной дофаминой терапии, не смогло обеспечить выход к первичной конечной точке, заявленной улучшением общего балла по унифицированной рейтинговой шкале оценки болезни Паркинсона Международного общества изучения двигательных расстройств (MDS-UPDRS), которая оценивает тяжесть и прогрессирование этого заболевания [4] [5].
Однако затем выяснилось, что улучшения всё же есть, и они связаны с моторными функциями: годичный курс терапии из ежемесячных вливаний прасинезумаба привел к 35-процентному относительно плацебо сдерживанию прогрессирования моторных нарушений, такие как замедленность движений (брадикинезия), тремор, ригидность, нестабильность походки — согласно изменению балла части III шкалы MDS-UPDRS [6] [7]. При этом благотворный эффект замедления ухудшений, оказавшийся справедливым главным образом среди лиц с быстрым прогрессированием болезни Паркинсона, продолжал усиливаться в ходе продолжения лечения сроком максимум 4 года [8].
В мае 2021 года было запущено клиническое исследование PADOVA (NCT04777331) фазы IIb, проверяющее, насколько хорошо прасинезумаб сдерживает процесс ухудшения моторных функций у пациентов (n=586) с болезнью Паркинсона в более запущенной, чем в PASADENA, форме. Результаты будут раскрыты к концу 2024 года.
В середине декабря 2024 года стало известно о провале PADOVA: прасинезумаб не смог статистически значимым образом опередить плацебо, хотя численно (номинально) превзошел, на 16% отсрочив время до подтвержденного прогрессирования ухудшения двигательных функций — отношение риска (hazard ratio, HR) 0,84 (95% ДИ [здесь и далее]: 0,69–1,01; p=0,0657). В популяции пациентов, получавших терапию леводопой (75% испытуемых), эффект был более выраженным: HR 0,79 (0,63–0,99; p=0,0431). Отмечены устойчивые положительные тенденции по множеству вторичных и эксплоративных конечных точек [9] [10].
Согласно анализу данных, скорректированных по ряду ковариат (включая возраст, пол, стадию по Хён и Яру, применение фоновых медикаментов), терапевтический эффект прасинезумаба оказался номинально значимым: соответственно HR 0,81 (0,67–0,98; p=0,0334) и HR 0,76 (0,61–0,95; p=0,0175).
МИНЗАСОЛМИН И UCB7853: UCB / NOVARTIS
В декабре 2021 года бельгийская «ЮСиБи» (UCB) договорились с «Новартис» (Novartis) о совместной разработке и коммерциализации двух лекарственных активов, предназначенных для лечения болезни Паркинсона [1].
По условиям соглашения,«ЮСиБи» получит от «Новартис» авансом $150 млн и последующие выплаты на сумму до $1,5 млрд долларов по мере прохождения определенных этапов развития экспериментальных проектов. В случае коммерциализации готовых лекарств доходы будут делиться территориально: «ЮСиБи» займется их реализацией в Европе и Японии, «Новартис» — в США и других странах.
Партнерство касается двух препаратов-кандидатов, нацеленных на альфа-синуклеин.
Минзасолмин (minzasolmin, UCB0599,NPT200-11, DLX-313), который «ЮСиБи» в 2014 году лицензировала у «Ньюропор терапис» (Neuropore Therapies), ингибирует внутриклеточную агрегацию альфа-синуклеина. Низкомолекулярный минзасолмин, будучи циклической пептидомиметической молекулой, является соединением второго поколения, оптимизированным для пероральной биодоступности и прохождения через гематоэнцефалический барьер.
Минзасолмин, который взаимодействует с C-концевым доменом альфа-синуклеина и предотвращает его связывание с мембранами с дальнейшей олигомеризацией в них, in vitro сдерживает агрегацию альфа-синуклеина, а на трансгенных мышиных моделях нормализует нейронные и воспалительные маркеры, устраняет моторный дефицит, ослабляет кортикальную альфа-синуклеиновую патологию и астроглиоз, нормализует уровень стриарного дофаминового транспортера [2] [3].
В декабре 2020 года «ЮСиБи» приступила к 18-месячному клиническому исследованию ORCHESTRA (NCT04658186) фазы IIa (рандомизированному, двойному слепому, плацебо-контролируемому, многоцентровому, международному), проверяющему терапевтическую эффективность минзасолмина среди пациентов (n=496) с болезнью Паркинсона на ранней стадии (стадия паркинсонизма ≤ 2,5 по Хён и Яру, длительность заболевания не дольше 2 лет). Результаты должны быть собраны ближе к концу 2024 года.
В середине декабря 2024 года стало известно о провале ORCHESTRA: минзасолмин не смог обеспечить выход ни к первичной конечной точке эффективности лечения болезни Паркинсона (изменение суммарного балла частей I–III шкалы MDS-UPDRS), ни к вторичным (изменения баллов отдельных частей этой шкалы) — экспериментальное лечение не замедлило прогрессирование заболевания [4].
Продолжается клиническое испытание NCT05543252 фазы II, изучающее долгосрочные безопасность, переносимость и эффективность минзасолмина при впервые поставленном диагнозе болезни Паркинсона пациентам (n=428) из других исследований этого экспериментального препарата. Оно завершится в 2027 году.
Отсутствие доказательств клинической пользы минзасолмина привело к прекращению NCT05543252.
UCB7853 — моноклональное антитело против альфа-синуклеина, подавляющее его внеклеточное распространение.
В декабре 2020 года было запущено клиническое исследование NCT04651153 фазы I, оценивающее различные внутривенные дозы UCB7853 с позиций безопасности, переносимости и фармакокинетики — среди здоровых добровольцев и пациентов с болезнью Паркинсона на легко-умеренной стадии. Испытание завершено, дальнейших шагов по развитию проекта пока не предпринималось.
В середине декабря 2024 года, после клинической неудачи минзасолмина, «ЮСиБи» подтвердила свою приверженность продолжению разработки UCB7853 [4].
В свое время утверждалось, что UCB0599 и UCB7853 органично дополняют друг друга в задаче замедления прогрессирования болезни Паркинсона: первый препарат показан на ранних ее стадиях, второй подходит при более запущенных состояниях.
Дофаминомиметик тавападон предназначен для улучшения моторных функций при паркинсонизме без обременяющих побочных эффектов.
ЭКСИДАВНЕМАБ: BIOARCTIC / ABBVIE
В руках «ЭббВи» (AbbVie) было гуманизированное моноклональное антитело эксидавнемаб (exidavnemab, ABBV-0805, BAN0805), в декабре 2018 года лицензированное у шведской «Байоарктик» (BioArctic) и связывающее олигомерные и протофибриллярные формы альфа-синуклеина различных конформаций с пикомолярной аффинностью [1] [2].
Эксидавнемаб характеризуется очень высокой (100 тыс. раз) селективностью связывания агрегированного альфа-синуклеина относительно его мономерных форм. Для сравнения: сродство прасинезумаба (prasinezumab, RG7935, PRX002) и цинпанемаба (cinpanemab, BIIB054), за которыми стоят «Рош» (Roche) / «Проутина» (Prothena) и «Байоджен» (Biogen) / «Ньюримьюн» (Neurimmune), к агрегированному альфа-синуклеину соответственно в 400 и 800 раз выше, чем к мономерному.
Учитывая высокий уровень физиологических мономеров альфа-синуклеина в крови, важно минимизировать его связывание, во-первых, во избежание периферической секвестрации антитела в плазме, чтобы его большее количество достигло своих мишеней в головном мозге, и, во-вторых, для улучшенного профиля безопасности и сниженной терапевтической дозы.
Согласно клинической проверке среди здоровых добровольцев (n=95), эксидавнемаб, назначаемый однократно внутривенно по 300–6000 мг или подкожно по 300 мг, сохраняет свою активность продолжительное время: период полувыведения составляет приблизительно 30 дней. Установлено дозозависимое снижение плазматической концентрации свободного альфа-синуклеина. Каких-либо проблем с безопасностью не выявлено [3].
Будущее эксидавнемаба казалось туманным, поскольку в июле 2020 года «ЭббВи» по стратегическим причинам остановила соответствующее клиническое испытание NCT04127695 фазы I. Впоследствии компания вышла из партнерства с «Байоарктик», которая стала самостоятельно развивать этот препарат-кандидат [4].
В ноябре 2024 года было организовано клиническое исследование EXIST (NCT06671938) фазы IIa среди пациентов (n=24) с легко-умеренной болезнью Паркинсона, следующих стабильным курсом симптоматической терапии. Завершение запланировано на 2026 год. По результатам «Байоарктик» примет окончательное решение, что делать с эксидавнемабом: сворачивать проект либо продолжать им заниматься в целях лечения болезни Паркинсона, деменции болезни Паркинсона, деменции с тельцами Леви или множественной системной атрофии.
PD01A: AC IMMUNE
Несмотря на скудные успехи игроков «Большой фармы», швейцарская «ЭйСи имьюн» (AC Immune) верит в разумность гипотезы альфа-синуклеина: для этого в июле 2021 года у австрийской «Аффирис» (AFFiRiS) была куплена PD01A — активная вакцина против альфа-синуклеина [1].
Иммуноген в составе PD01A представляет собой пептид из восьми аминокислот (PD01), который имитирует эпитоп в C-концевой области человеческого альфа-синуклеина, но с другой аминокислотной последовательностью. Пептид конъюгирован с белком-носителем гемоцианином лимфы улитки (KLH) и адсорбирован на адъювантном гидроксиде алюминия. Вакцина PD01A разработана таким образом, чтобы стимулировать В-клеточный антительный ответ, но обходить аутореактивную мобилизацию Т-клеток, которая может вызвать ненужные нейровоспалительные реакции.
Активная иммунизация при помощи PD01A генерирует антитела, которые избирательно распознают агрегаты альфа-синуклеина с гораздо меньшим сродством к его мономерным формам и без реактивности к бета-синуклеину. Белок-носитель обеспечивает необходимые эпитопы Т-хелперов для индукции длительного и усиленного ответа антител, тогда как антигенный компонент (PD01) действует исключительно как В-клеточный эпитоп и отвечает за специфичность гуморального иммунного ответа [2].
В июле 2023 года «ЭйСи» запустила клиническое испытание VacSYn (NCT06015841) фазы II, которое изучает экспериментальную вакцину ACI-7104.056, представляющую собой PD01 в оптимизированной рецептуре, среди пациентов (n=150) с болезнью Паркинсона на начальной стадии. К концу 2024 года будет предоставлен первый промежуточный отчет о безопасности и иммуногенности вакцины. Срок окончания исследования — начало 2028 года.
Ничего не получилось у «Байоджен» (Biogen) с моноклональным антителом цинпанемаб (cinpanemab, BIIB054), лицензированным у швейцарской «Ньюримьюн» (Neurimmune) и таргетированным против агрегатов альфа-синуклеина. Хотя в отрасли считалось, что этот препарат наиболее совершенный с технической точки зрения. В клиническом испытании SPARK (NCT03318523) фазы II цинпанемаб не обеспечил каких-либо значимых улучшений по шкале MDS-UPDRS. В феврале 2021 года дальнейшая разработка цинпанемаба была прекращена [1].
ABL301: ABL BIO / SANOFI
В середине января 2022 года «Санофи» (Sanofi) приобрела у корейской «Эй-би-эл байо» (ABL Bio) мировую лицензию на экспериментальный препарат ABL301 (SAR446159), предназначенный для лечения болезни Паркинсона [1].
Сделка предполагает выплату $75 млн авансом и последующей суммы до $985 млн по мере развития проекта, а также роялти от продаж готового лекарства.
ABL301 представляет собой биспецифическое моноклональное антитело, таргетированное на альфа-синуклеин и рецептор инсулиноподобного фактора роста 1 (IGF1R) [2] [3].
ABL301 с высокой аффинностью распознаёт патологические агрегаты альфа-синуклеина, обходя стороной его мономерные формы. Нацеливание на IGF1R, сделанное в рамках технологической платформы Grabody-B, позволяет ABL301 без проблем миновать гематоэнцефалический барьер (ГЭБ). Поскольку IGF1R высоко и относительно специфично экспрессирует на эндотелиальных клетках головного мозга, ABL301 проникает в мозг посредством рецептор-опосредованного трансцитоза (RMT).
Как утверждает «Эй-би-эл», ее ABL301 характеризуется рядом выгодных преимуществ перед другими моноклональными антителами, которые разрабатывают игроки фармотрасли. Во-первых, биологические лекарственные препараты конкурентов весьма плохо проникают через ГЭБ: в головном мозге обнаруживается приблизительно 0,1–0,2% таких антител.
Во-вторых, биопрепараты соперников фактически без особого разбора связывают агрегированные и мономерные формы альфа-синуклеина, хотя именно первые вовлечены в патофизиологические каскады при болезни Паркинсона.
В-третьих, ABL301 располагает двойным механизмом действия: содействие фагоцитозу внеклеточного альфа-синуклеина микроглиальными клетками и подавление прионоподобного распространения альфа-синуклеина от клетки к клетке. Антитела конкурентов в основном осуществляют только второе.
Продолжается клиническое испытание NCT05756920 фазы I среди здоровых добровольцев (n=86). Его завершение намечено к началу 2025 года.
БУНТАНЕТАП: ANNOVIS BIO
Интересным видится подход «Анновис байо» (Annovis Bio): ее низкомолекулярный препарат-кандидат бунтанетап (buntanetap, ANVS401), также известный как посифен (posiphen), нацелен сразу на три нейротоксичных белка — бета-амилоид, альфа-синуклеин и тау-белок. Все они нарушают аксональный транспорт нейромедиаторов и нейротрофических факторов и замедляют синаптическую передачу, тем самым ухудшая нервную деятельность в целом. Подобные нарушения приводят к активации иммунной системы, которая атакует нервные клетки, что приводит к нейровоспалению, дегенерации и смерти нервных клеток. Итогом становится ухудшение когнитивной и моторной деятельности.
Клиническое исследование NCT04208152 фазы I протестировано эмрусолмин среди взрослых добровольцев (n=68), установив его приемлемые безопасность и переносимость [1].
Клиническое испытание NCT04685265 фазы Ib проверило назначение различных доз эмрусолмина пациентам с легко-умеренной болезнью Паркинсона. Исследование, завершившееся в конце 2022 года, результатов так и не предоставило.
В конце октября 2021 года к проекту эмрусолмина подключилась «Тева фармасьютикал индастриз» (Teva Pharmaceutical Industries), пожелавшая обзавестись мировыми правами на этот препарат-кандидат [2]. Партнерство также касается sery433 — пролекарства эмрусолмина, разработанного с прицелом повышения его биодоступности [3].
«Тева» осуществляет 56-недельное клиническое испытание TOPAS-MSA (NCT06568237) фазы II, изучающее эмрусолмин в лечении множественной системной атрофии. Результаты будут собраны к середине 2027 года.
Пероральный низкомолекулярный эмрусолмин, эффективно преодолевающий гематоэнцефалический барьер (ГЭБ), специфически и с наномолярной аффинностью связывает токсичные олигомерные структуры внутриклеточного альфа-синуклеина, что приводит к их растворению и предотвращению образования новых олигомеров.
Поскольку эмрусолмин разрушает преамилоидные олигомеры и нарушает рост фибрилл, это приводит, в сочетании с механизмами клеточного клиренса, к уменьшению количества отложенных амилоидных фибрилл. Эмрусолмин не является разрушителем фибрилл, что важно, так как их фрагментация усиливает прионоподобное распространение токсичных белков и прогрессирование болезни за счет образования большего количества частиц, способных привлекать мономеры путем неправильного фолдинга.
Эмрусолмин, будучи дифенилпиразольным соединением, подавляет образование олигомеров альфа-синуклеина и прионного белка. На различных мышиных моделях синуклеопатий и болезни Паркинсона эмрусолмин препятствовал накоплению олигомеров и дегенерации нейронов [4], улучшил функцию дофаминовых нейронов и моторные функции [5], сдержал прогрессирование заболевания, причем даже после клинической манифестации болезни Паркинсона [6].
Утверждается также, что поскольку патологические олигомеры при нейродегенеративных заболеваниях имеют общие структурные особенности, хотя основной белковый компонент специфичен для каждой болезни, а эмрусолмин модулирует образование олигомеров путем воздействия на структурно-зависимые эпитопы, эта молекула, не исключено, может найти себя в лечении различных патологий, связанных с агрегацией белков [4].
На мышиных моделях множественной системной атрофии эмрусолмин снизил количество олигомеров альфа-синуклеина, сохранил дофаминергические нейроны и улучшил ходьбу [7], сдержал нейродегенерацию в черном веществе и уменьшил микроглиальную активацию [8]. На модели, имитирующей более тяжелой форму этого заболевания, эмрусолмин несколько улучшил двигательные навыки, но существенно не повлиял на нейродегенерацию или накопление альфа-синуклеина [9].
На мышиных моделях тау-патологии эмрусолмин уменьшил потерю нейронов, улучшил когнитивные способности и продлил выживаемость [10], причем даже у старых мышей он смог обратить вспять нарушения конечного метаболизма глюкозы в головном мозге [11].
На мышиной модели болезни Альцгеймера эмрусолмин восстановил синаптическую пластичность гиппокампа и память [12].
АМБРОКСОЛ
Амброксол (ambroxol) —муколитическое лекарственное средство и основной ингредиент ряда безрецептурных противокашлевых препаратов. В США и Канаде амброксол не зарегистрирован.
Интерес к изучению амброксола в лечении болезни Паркинсона обусловлен его активностью в качестве молекулярного шаперона для лизосомального фермента бета-глюкоцереброзидазы (GCase): мутации с потерей функции в гене GCase (GBA1) ассоциированы с повышенным в 20–30 раз риском развития болезни Паркинсона, причем с более ранним началом и ускоренными когнитивными и моторными ухудшениями [1] [2].
Активность бета-глюкоцереброзидазы и альфа-синуклеина связаны между собой: дефицит GCase вызывает патологическое накопление альфа-синуклеина в клеточных культурах [3], а сверхэкспрессия GCase в головном мозге ослабляет патологию и дефицит памяти в мышиной модели синуклеинопатии [4]. Активность GCase снижена при идиопатической болезни Паркинсона без мутаций GBA1 [5], снижение активности GCase коррелирует с более ранним началом заболевания и ухудшением когнитивных и немоторных симптомов [6]. Мутации GBA1 были первоначально идентифицированы [7] как причина [8] паркинсонизма при болезни Гоше — лизосомальном заболевании, характеризующемся накоплением сфинголипидов и в некоторых случаях патологией альфа-синуклеина.
В клетках, полученных от пациентов с болезнью Гоше или болезнью Паркинсона, амброксол стабилизировал мутантную GCase [9] и способствовал ее перемещению из эндоплазматического ретикулума (ER) в лизосомы [10], повышая уровень белка, активность фермента и функцию лизосом. У мух с мутациями GCH1 амброксол усиливал активность GCase, снижал стресс ER и защищал двигательную функцию [11].
У мышей дикого типа и мышей с мутацией GCH1 амброксол индуцировал активность GCase в головном мозге, а у мышей, сверхэкспрессирующих человеческий альфа-синуклеин, снижал общее количество и уровень фосфорилированного альфа-синуклеина [12]. В мышиной модели бокового амиотрофического склероза амброксол улучшал двигательную функцию и продлевал выживаемость [13].
У здоровых нечеловекообразных приматов ежедневный прием амброксола усиливал активность GCase в головном мозге [14].
Клиническое испытание AiM-PD (NCT02941822) фазы II, проведенное Университетским колледжем Лондона (University College London, UCL, Лондон, Великобритания), продемонстрировало, что ежедневные 1260 мг амброксола (многократно увеличенная доза, применяемая при кашле), назначаемые в течение пяти месяцев, способствовали улучшению показателей по шкале MDS-UPDRS (в особенности моторных функций) у пациентов с болезнью Паркинсона умеренной тяжести [15].
Лоусоновский научно-исследовательский институт здоровья (Lawson Health Research Institute, London, Онтарио, Канада) осуществляет клиническую проверку NCT02914366 фазы II на предмет того, сможет ли амброксол улучшить когнитивные и моторные симптомы при деменции на фоне болезни Паркинсона. Сроки завершения установлены на конец 2025 года [16].
Анализ реальных данных пациентов с болезнь Гоше, в том числе страдающих болезнью Паркинсона на ее фоне, которые принимали амброксол, подтвердил оказываемые им благотворные эффекты, проявляющиеся стабилизацией или улучшением неврологического статуса, увеличением физической активности, снижением утомляемости [17].
ВЕКТОРИЗИРОВАННЫЕ АНТИТЕЛА: VOYAGER THERAPEUTICS
Интересную идею продвигает «Вояджер терапьютикс» (Voyager Therapeutics), придумавшая решение проблемы доставки в головной мозг необходимого количества антител, связывающих патологические белки, в том числе альфа-синуклеин. «Вояджер» занялась вопросом обхождения гематоэнцефалического барьера (ГЭБ), препятствующего проникновению крупных лекарственных молекул. Да, можно осуществлять частые системные инъекции антител, но это не выход, поскольку высока вероятность побочных реакций.
Принцип так называемых векторизированных антител следующий: конструируется аденоассоциированный вирусный вектор (AAV), который несет определенные генетические инструкции, кодирующие нужное моноклональное антитело, синтез которого осуществляется самим организмом прямиком в головном мозге. Заявлено, что однократной внутривенной инъекции AAV-препарата окажется достаточно для оказания должного терапевтического эффекта очень продолжительное время — возможно, пожизненно. Объясняется это тем, что клетками-мишенями в центральной нервной системы в данном случае являются нейроны, долгоживущие и неделящиеся [1].
Всё бы ничего, но в августе 2020 года «ЭббВи» (AbbVie)разорвала с «Вояджер» мощное партнерское соглашение по векторизированным антителам против нейродегенеративных заболеваний.
ПРИРОДНЫЕ ВЕЩЕСТВА
Природные вещества (ПВ) — это соединения, полученные из природных источников, таких как растения, животные и микроорганизмы. С развитием современной химии и фармакологии выделение и характеристика ПВ становятся всё более сложными, открывая новые возможности для поиска лекарств. Многие ПВ обладают значительной биологической активностью и проявляют многообещающие терапевтические эффекты при широком спектре заболеваний.
Определенные природные вещества обладают способностью подавлять агрегацию альфа-синуклеина [1], однако клинических исследований, которые бы безговорочно подтвердили это чрезвычайно мало. Поэтому приходится полагаться либо на теоретические изыскания, либо на ограниченные данные применения ПВ при болезни Паркинсона.
Большую группу среди таких противопаркинсонических ПВ занимают полифенольные соединения: куркумин (curcumin) и его аналоги и производные, ресвератрол (resveratrol), пицеатаннол (piceatannol), катехины зеленого чая, гесперетин (hesperetin) и гесперидин (hesperidin), дигидромирицетин (dihydromyricetin, DHM) и сальвианоловая кислота B (salvianolic acid B, Sal B), рутин (rutin), текторигенин (tectorigenin), байкалеин (baicalein).
Способностью противодействовать нейротоксичным альфа-синуклеиновым агрегатам обладают следующие природные вещества: нафтохиноны (шиконин [shikonin], витамин K), таншиноны, экстракты грибов Ganoderma spp. («Линчжи», «Рейши», «Гриб бессмертия»), экстракты центеллы азиатской (Centella asiatica), производные коричной кислоты (cinnamic acid), экстракты гравилата городского (Geum urbanum), (+)-десдиметилпинорезинол [(+)-desdimethylpinoresinol], алкалоиды (хинолины и индолы, никотин, кофеин, скваламин [squalamine], тродускумин [trodusquemine]), а также пирролохинолинхинон (pyrroloquinoline quinone).
Болезнь Паркинсона — распространенное и изнуряющее нейродегенеративное заболевание.
Никакое из лекарств не в силах остановить неуклонное прогрессирование болезни Паркинсона, сопровождающееся нарастающей инвалидизацией.
GLP1R-агонисты, вовсю применяемые в лечении сахарного диабета и ожирения, открылись с неожиданной стороны.
Собраны многочисленные доказательства, что препараты вроде семаглутида обладают нейропротекторным действием, сдерживающим ухудшение двигательных функций при болезни Паркинсона.
Параллельно GLP1R-агонисты изучаются в лечении неалкогольного стеатогепатита (НАСГ), болезни Альцгеймера, инсульта, алкоголизма.
ЧТО ПРОИЗОШЛО
Добавление препарата «Адликсин» / «Ликсумия» (Adlyxin / Lyxumia, ликсисенатид) к стандартной терапии болезни Паркинсона привело к замедлению процесса ухудшения двигательных (моторных) функций, таких как дрожание конечностей (тремор), медлительность и скованность движений, трудности с удержанием равновесия.
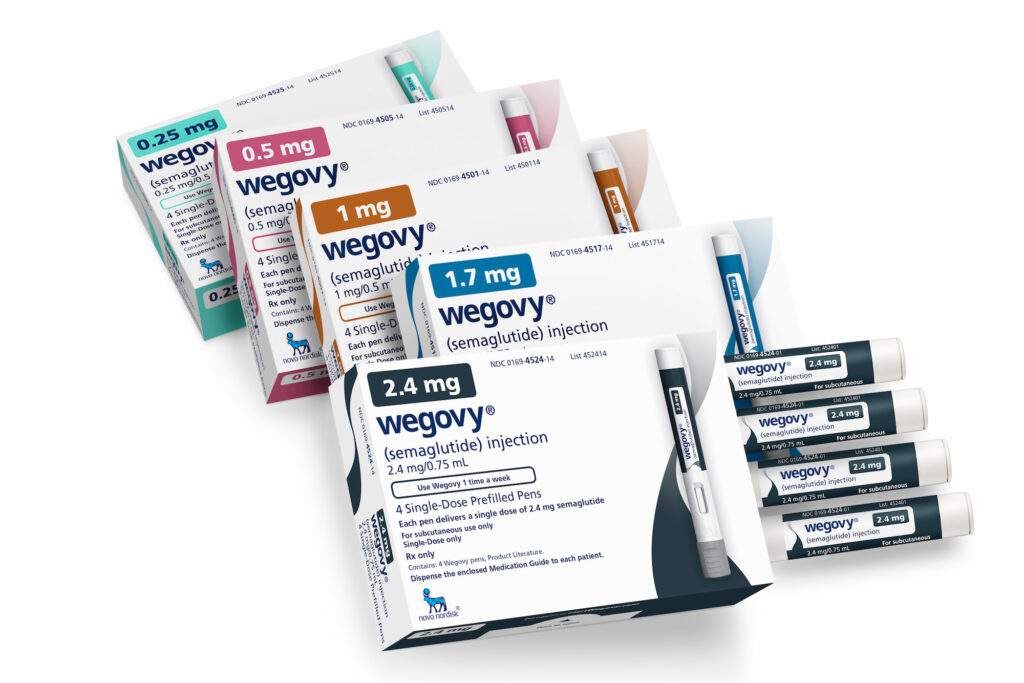
Ликсисенатид (lixisenatide) относится к классу агонистов рецептора глюкогоноподобного пептида 1 (GLP1R) — такому же, к которому принадлежат мегапопулярные «Оземпик» (Ozempic, семаглутид), «Вегови» (Wegovy, семаглутид), «Мунджаро» (Mounjaro, тирзепатид) и «Зепбаунд» (Zepbound, тирзепатид), разработанные «Ново Нордиск» (Novo Nordisk) и «Илай Лилли» (Eli Lilly) и вовсю применяемые в лечении сахарного диабета 2-го типа и ожирения.
В России семаглутид, защищенный патентами оригинатора до 2035 года, но в конце 2023 года получивший принудительную лицензию на производство, доступен в виде следующих препаратов: «Семавик» (Semavic), «Квинсента» (Queensenta) и «Инсудайв» (Insudive). Эти генерические лекарственные средства никак и ничем не уступают брендовым препаратам, притом что стоят существенно дешевле. Тирзепатид пока не зарегистрирован, но соответствующая клиническая проверка проводится; не исключено, он появится в виде недорогого дженерика.
«Адликсин» / «Ликсумия», в свое время продвигавшийся «Санофи» (Sanofi), более не реализуется: французский фармацевтический гигант от него отказался ввиду наличия более совершенных и удобных в использовании GLP1R-агонистов других фармпроизводителей, включая вышеперечисленные.
Болезнь Паркинсона — распространенное, изнуряющее и инвалидизирующее нейродегенеративное заболевание. Тремор в покое, ригидность конечностей, медлительность движений — вот ее наиболее известные признаки, которым сопутствуют осложнения в виде вегетативных симптомов, нарушения сна и когнитивных расстройств. Неуклонное прогрессирование болезни Паркинсона отражается постепенно нарастающей инвалидизацией, справиться с которой не под силу никакому из существующих фармакологических методов лечения [1]. Разработка нейропротекторных методов лечения, способных замедлить, остановить или обратить вспять нейродегенерацию при болезни Паркинсона, уже давно является приоритетной задачей [2].
В 1817 году Джеймс Паркинсон (James Parkinson) в своем «Эссе о дрожательном параличе» оптимистично писал, что, хотя природа болезни ему неизвестна, «существуют достаточные основания надеяться, что в скором времени будет открыт некий лечебный процесс, с помощью которого, по крайней мере, можно будет остановить прогрессирование болезни» [3]. Прошло более двухсот лет, а мы всё еще ждем этого открытия.
Бунтанетап действует сразу на три нейротоксичных белка, ответственных за нейродегенеративные нарушения, — бета-амилоид, альфа-синуклеин и тау-белок.
ЧТО ВЫЯСНИЛОСЬ
Клиническое исследование LixiPark (NCT03439943) фазы II (рандомизированное, двойное слепое, плацебо-контролируемое, многоцентровое) охватило французских пациентов (n=156) в возрасте 40–75 лет с ранней болезнью Паркинсона (стадия < 3 по Хён и Яру), которые придерживались стандартной противопаркинсонической дофаминергической терапии и которым дополнительно на протяжении 12 месяцев ежедневно назначали подкожные инъекции ликсисенатида (lixisenatide) или плацебо.
Первичная конечная точка эффективности лечения была установлена изменением балла в части III унифицированной рейтинговой шкалы оценки болезни Паркинсона Международного общества изучения двигательных расстройств (MDS-UPDRS III). В группе ликсисенатида это изменение составило −0,04 балла, свидетельствуя об улучшении моторных функций, — против +3,04 балла, указывая на ухудшение статуса инвалидизации. Статистически значимая разница составила 3,08 балла (95% ДИ [здесь и далее]: 0,86–5,30; p=0,007) [1].
По прошествии 2-месячного отмывочного периода расхождение сохранилось, причем даже в те моменты, когда пациенты не принимали никаких противопаркинсонических препаратов: усредненный балл получился равным 17,7 пункта (15,7–19,7) — против 20,6 (18,5–22,8) [меньше — лучше].
Применение ликсисенатида сопровождалось тошнотой и рвотой у 46% и 13% испытуемых соответственно.
СУТЬ
GLP1R-агонист ликсисенатид оказал умеренно выраженное благотворное влияние на сдерживание прогрессирования двигательной инвалидизации при болезни Паркинсона, что, возможно, связано с оказываемым им нейропротекторным действием. Предположительно, положительное действие ликсисенатида особенно хорошо себя проявит при возрасте не старше 60 лет и заболевании на относительно ранней стадии. Впрочем, нельзя исключать симптоматического эффекта: доклиническая и клиническая проверка GLP1R-агониста эксенатида (exenatide) при алкогольной и кокаиновой зависимости показала, что он повышает уровень синаптического дофамина [1] [2].
ОДНАКО
Ограниченность клинического испытания лечения болезни Паркинсона GLPR1-агонистом ликсисенатидом не позволяет установить, сохранится ли оказываемый на моторные функции положительный эффект препарата при более длительном его применении или более тяжелой форме заболевания. Неизвестна также величина эффекта большей или меньшей дозы ликсисенатида.
КАК ЭТО РАБОТАЕТ
На данном этапе существуют лишь догадки, что GLP1R-агонисты помогают в лечении болезни Паркинсона, если судить по набору релевантных научных наработок. Так, сахарный диабет 2-го типа является фактором риска развития болезни Паркинсона [1], а его лечение GLP1R-агонистами ассоциировано со снижением этого риска более чем на 50% [2]. На животных моделях болезни Паркинсона GLP1R-агонисты продемонстрировали нейропротекторное действие [3]. В ответ на активацию GLP1R наблюдаются различные физиологические эффекты, в том числе уменьшение воспаления в головном мозге — процесса, который занимает центральной место в патофизиологии болезни Паркинсона [4]. Есть мнение, что активация GLP1R стимулирует нейрогенез и защищает нейроны от опосредованного цитокинами апоптоза, в том числе путем предотвращения микроглиального преобразования астроцитов в нейротоксичный фенотип [3] [5].
Дофаминомиметик тавападон предназначен для улучшения моторных функций при паркинсонизме без обременяющих побочных эффектов.
ЧТО ДАЛЬШЕ
Большинство пациентов с болезнью Паркинсона беспокоит не их нынешнее состояние, а страх прогрессирования моторных нарушений. Если улучшение двигательных функций на 3 балла по шкале MDS-UPDRS III — тот максимум, которого можно добиться от GLP1R-агонистов, то ценность подобного лечения незначительна, особенно с учетом обременительных нежелательных явлений со стороны желудочно-кишечного тракта. Однако если польза такой терапии проявит кумулятивный, накопительный характер, к примеру, добавляя по 3 балла каждый год в течение 5–10 лет и дольше, тогда можно смело говорить о появлении первого в мире лечения, преобразующего и изменяющего течение болезни Паркинсона. Необходимы соответствующие долгосрочные клинические испытания [1].
В БЛИЖАЙШЕМ БУДУЩЕМ
Во второй половине 2024 года ситуация с лечением болезни Паркинсона при помощи GLP1R-агонистов станет более ясной, когда будут готовы результаты клинического исследования Exenatide-PD3 (NCT04232969) фазы III, в котором GLP1R-агонист эксенатид, коммерциализированный «АстраЗенека» (AstraZeneca) как противодиабетический «Бидуреон» (Bydureon), на протяжении 2 лет назначается еженедельными подкожными инъекциями пациентам (n=194) в возрасте 25–80 лет с ранней болезнью Паркинсона (стадия ≤ 2,5 по Хён и Яру), придерживающихся стандартной противопаркинсонической терапии [1].
Итоги Exenatide-PD3 оказались разочаровывающими: не обнаружено статистически значимой разницы между эксенатидом и плацебо в том, что касается сдерживания прогрессирующего ухудшения моторных функций при болезни Паркинсона [2]. Исследователи Университетского колледжа Лондона (University College London, UCL, Лондон, Великобритания) продолжают выяснять, почему эксенатид не справился с поставленной задачей — вопреки ликсисенатиду, который был эффективен.
РАНЕЕ
Предшествовавшие клинические исследования эксенатида, а также экспериментального NLY01, пегилированной версии эксенатида авторства «Ньюрали» (Neuraly), выдали неоднозначные результаты лечения болезни Паркинсона, зависящие от особенностей пациентов [1] [2] [3].
Фармацевтическая отрасль продолжает упорную борьбу с распространенным нейродегенеративным заболеванием.
И ЕЩЁ
Южнокорейская «Пептрон» (Peptron) вынашивала грандиозные планы на экспериментальный PT320 — рецептуру эксенатида с замедленным высвобождением, которая сделана по фирменной технологии SmartDepot на базе биоразлагаемых полимерных микросферических носителей и которая позволяет назначать препарат подкожными инъекциями один раз в две недели [1] [2] [3] [4]. Однако клиническое испытание NCT04269642 фазы II среди пациентов с ранней болезнью Паркинсона, начатое весной 2020 года, так и не завершилось.
ТЕМ ВРЕМЕНЕМ
GLP1R-агонисты продолжают демонстрировать свою пользу за пределами исключительно сахарного диабета 2-го типа и ожирения.
Так, продвигаемый «Ново Нордиск» (Novo Nordisk) семаглутид (semaglutide) доказал, что, во-первых, снижает риск неблагоприятных исходов при сердечно-сосудистом заболевании на фоне ожирения и, во-вторых, успешно справляется с лечением сердечной недостаточности с сохраненной фракцией выброса (HFpEF) среди пациентов с ожирением. Впрочем, это было предсказуемо, учитывая, насколько лишний вес токсичен для сердечно-сосудистой системы.
Эффективное снижение веса при помощи семаглутида сопровождается снижением риска сердечно-сосудистой смерти, инфаркта миокарда и инсульта.
Семаглутид также смог сдержать прогрессирование хронической болезни почек и снизить риск сердечно-сосудистой и почечной смерти у пациентов с сахарным диабетом 2-го типа [1].
Летом 2024 года «Илай Лилли» (Eli Lilly) расскажет, насколько «Зепбаунд» (Zepbound, тирзепатид) терапевтически востребован в случае HFpEF с сопутствующим ожирением: этот вопрос раскрывается в клиническом исследовании SUMMIT (NCT04847557) фазы III.
К осени 2025 года станет известно, пригоден ли семаглутид в лечении болезни Альцгеймера: способен ли «Ребелсас» (Rybelsus), принимаемый ежедневно перорально на протяжении 2 лет в рамках клинического испытания EVOKE (NCT04777396) фазы III, замедлить прогрессирование деменции у пациентов с ранней болезнью Альцгеймера.
Семаглутид против ожирения попутно ослабит бремя сердечной недостаточности с сохраненной фракцией выброса.
Весной 2026 года ожидаются результаты клинического исследования GALLOP (NCT05920889) фазы II, которое проверяет гипотезу, что добавление семаглутида к стандартной механической процедуре эндоваскулярной тромбэктомии (EVT) при остром ишемическом инсульте, вызванном окклюзией крупных сосудов, предотвращает неблагоприятные исходы, обусловленные перипроцедурным злокачественным отеком мозга (MBE) и симптоматическим внутричерепным кровоизлиянием (sICH).
Тема неалкогольного стеатогепатита (НАСГ), интересующая буквально каждого игрока «Большой фармы» ввиду огромных бизнес-перспектив, но по факту остающаяся без действительно сильных лекарств, прорабатывается в клиническом исследовании ESSENCE (NCT04822181) фазы III, в котором изучается длительное (максимум 5 лет) еженедельное применение инъекционного семаглутида при НАСГ без цирроза печени и с ее фиброзом на стадии F2–F3.
Продолжается тестирование семаглутида среди людей с коморбидным ожирением и алкогольной зависимостью: по силам ли «Вегови» (Wegovy), который в ходе клинического исследования SEMALCO (NCT05895643) фазы II назначается еженедельными подкожными инъекциями, снизить потребление алкоголя или даже избавить от алкоголизма.
Экспериментальный препарат бепировирсен (bepirovirsen), разрабатываемый «ГлаксоСмитКляйн» (GlaxoSmithKline), располагает необходимым потенциалом, для того чтобы обеспечить функциональное излечение хронического вирусного гепатита B.
ОСНОВНЫЕ ФАКТЫ
Продемонстрировано, что еженедельные подкожные инъекции бепировирсена привели к полному клиренсу ДНК и поверхностного антигена вируса гепатита В (HBsAg) у почти трети пациентов после 6 месяцев лечения.
Впрочем, по прошествии 6 месяцев наблюдений после завершения терапии статус функционального излечения оказался справедливым для существенно меньшей пропорции больных. Однако терапию всё равно можно назвать успешной, если сравнивать с существующими лекарственными средствами.
В настоящее время хронический вирусный гепатит B, которым заражены 254 млн человек во всём мире, неизлечим: приходится придерживаться пожизненной терапии, которая далеко не всегда оказывается успешной [1].
Напротив, хронический вирусный гепатит C считается полностью излечимым заболеванием. С мая 2011 года стали планомерно появляться всё более эффективные противовирусные препараты прямого действия (ПППД), за несколько месяцев исцеляющие эту инфекционную болезнь.
Цена лекарств постоянно растет, несмотря на отсутствие значимых улучшений. Разбираемся, почему фармацевтическая индустрия превратилась в машину для выкачивания денег.
КАК ЭТО РАБОТАЕТ
Бепировирсен (bepirovirsen, GSK3228836, IONIS-HBVRx) представляет собой антисмысловой олигонуклеотид (ASO) — одноцепочечную ДНК, которая комплементарна 20 консервативным нуклеотидным последовательностям всех матричных РНК (мРНК) вируса гепатита B (HBV), включая его прегеномную РНК (пгРНК).
Связывание бепировирсена с мРНК и пгРНК HBV приводит к образованию гибридного комплекса, который рекрутирует эндогенную рибонуклеазу H (РНКаза H). Этот фермент расщепляет мРНК и пгРНК HBV, тем самым препятствуя трансляции белков вируса. В результате уменьшается количество РНК, ДНК и белков HBV, в том числе поверхностного антигена вируса гепатита В (HBsAg). Вирусная нагрузка снижается, сдерживаются процессы инфицирования и репликации HBV, появляется шанс на функциональное излечение хронического вирусного гепатита B [1] [2].
Бепировирсен также проявляет агонистическую активность в отношении толл-подобного рецептора 8 (TLR8), тем самым работая как иммуностимулятор, индуцирующий активность врожденного иммунитета и выработку цитокинов [3] [4] [5] [6] [7].
Поскольку бепировирсен не конъюгирован с аминосахаром N-ацетилгалактозамином (GalNAc), нужным для более таргетной доставки в печень, он в большей степени распределяется в непаренхимных клетках печени, нежели в гепатоцитах, и потому распознается резидентными иммунными клетками печени, что приводит к активации сигнализации врожденной иммунной системы [8].
Бепировирсен разработан «Айонис фармасьютикалс» (Ionis Pharmaceuticals), которая в конце августа 2019 года лицензировала его «ГлаксоСмитКляйн» (GlaxoSmithKline). Взамен обещано до 262 млн долларов и роялти от реализации готового лекарственного препарата [9].
Состоятельность терапевтической парадигмы антисмысловых олигонуклеотидов подтверждена немалым числом уже одобренных лекарственных препаратов, направленных на сдерживание экспрессии специфических белков, патогенных в случае какого-либо заболевания. Так, например, «Эксондис 51» (Exondys 51, этеплирсен), «Виондис 53» (Vyondys 53, голодирсен), «Амондис 45» (Amondys 45, касимерсен) и «Вилтепсо» (Viltepso, вилтоларсен) применяются в лечении мышечной дистрофии Дюшенна, «Спинраза» (Spinraza, нусинерсен) используется в лечении спинальной мышечной атрофии, «Тегседи» (Tegsedi, инотерсен) назначается для лечения полинейропатии при наследственном транстиретиновом амилоидозе.
КЛИНИЧЕСКИЕ ПОДРОБНОСТИ
В целях предварительного выяснения эффективности и безопасности лечения хронического вирусного гепатита B при помощи бепировирсена «ГлаксоСмитКляйн» положилась на клиническую программу из трех испытаний фазы II:
B-Clear (NCT04449029): 24- или 12-недельное назначение бепировирсена (разными схемами), причем либо на фоне терапии нуклеозидными/нуклеотидными аналогами (NA), либо без нее.
B-Together (NCT04676724): 24- или 12-недельное применение бепировирсена на фоне NA-терапии, за которым следует 24-недельный курс пегилированного интерферона альфа-2a.
Первичная конечная точка эффективности лечения хронического вирусного гепатита B в первых двух клинических испытаниях была установлена пропорцией пациентов, продемонстрировавших устойчивую вирусную супрессию (подавление вирусной нагрузки; SVR), под которой понимают снижение уровней поверхностного антигена вируса гепатита В (HBsAg) и ДНК HBV ниже определяемых высокоточным методом ПЦР порогов (соответственно 0,05 МЕ/мл и 20 МЕ/мл), сохраняющееся на протяжении 24 недель после завершения лечения и при условии отсутствия в этот период дополнительной терапии какими-либо противовирусными препаратами (если таковые ранее не принимались).
B-Sure (NCT04954859): среди ранее прошедших лечение бепировирсеном и ответивших на него пациентов из других клинических испытаний. В ходе 33-месячных наблюдений выясняется, как долго сохраняется SVR-статус.
B-CLEAR
Клиническое исследование B-Clear (NCT04449029) фазы IIb (рандомизированное, двойное слепое, плацебо-контролируемое, многоцентровое, международное) пригласило взрослых пациентов (n=457) с хроническим вирусным гепатитом B, проходящих либо нет фоновое лечение нуклеозидными/нуклеотидными аналогами (NA).
На протяжении 24 недель участникам еженедельно подкожными инъекциями назначали бепировирсен (разными схемами):
группа 1: бепировирсен 300 мг — 24 недели;
группа 2: бепировирсен 300 мг — 12 недель, затем бепировирсен 150 мг — 12 недель;
группа 3: бепировирсен 300 мг — 12 недель, затем плацебо — 12 недель;
группа 4: плацебо — 12 недель, затем бепировирсен 300 мг — 12 недель.
Группы 1, 2 и 3 также получили нагрузочные дозы бепировирсена: по 300 мг на 4-й и 11-й дни.
Согласно промежуточному анализу собранных данных, после 24-недельной терапии хронического вирусного гепатита B наилучшую результативность показала группа 1. Так, одновременное отсутствие HBsAg и ДНК HBV зафиксировано для 28% и 29% испытуемых, соответственно придерживавшихся фоновой терапии NA и не принимавших такие препараты. При этом у 68% и 65% участников уровень HBsAg упал ниже 100 МЕ/мл [1].
Итоговые результаты клинического исследования получились следующими [2].
Среди тех пациентов, которые придерживались фоновой терапии NA, к первичной конечной точке эффективности лечения вышли 9%, 9%, 3% и 0% участников в группах 1, 2, 3 и 4. Среди тех, кто не получал NA, первичная конечная точка достигнута среди 10%, 6%, 1% и 0% испытуемых.
Если допустить «всплески» активности вируса гепатита B (однократное повышение HBsAg или ДНК HBV до уровней, превышающих или равных пороговым), то есть несколько ослабить критерии выхода к первичной конечной точке, ее достигли 10%, 9%, 4% и 2% пациентов на фоновой терапии NA и 14%, 6%, 1% и 4% пациентов без фоновой терапии NA.
Успешный ответ на назначение бепировирсена напрямую зависел от исходного уровня HBsAg до начала лечения: он чаще фиксировался у испытуемых с низким уровнем HBsAg (≤ 3 log10 МЕ/мл) и реже с высоким (> 3 log10 МЕ/мл). К примеру, в группе 1 первичная конечная точка была засвидетельствована у 16% и 25% пациентов с низким изначальным уровнем HBsAg и у 6% и 7% пациентов с высоким — соответственно среди получавших и не получавших фоновую терапию NA.
Бепировирсен характеризовался приемлемой переносимостью. Наиболее распространенным нежелательным явлением были реакции по месту введения препарата.
B-TOGETHER
Клиническое исследование B-Together (NCT04676724) фазы IIb (рандомизированное, открытое, многоцентровое, международное) пригласило взрослых пациентов (n=108) с хроническим вирусным гепатитом B.
Испытуемым, продолжающим следовать стабильной терапии нуклеозидными/нуклеотидными аналогами (NA), назначали бепировирсен (еженедельными подкожными 300-мг инъекциями) на протяжении 24 недель (группа 1) или 12 недель (группа 2); плюс нагрузочные дозы бепировирсена (по 300 мг на 4-й и 11-й дни). Далее участники проходили 24-недельный курс пегилированного интерферона альфа-2a (в еженедельной подкожной дозе 180 мкг).
Экспериментальное лечение обеспечило выход к первичной конечной точке для 9% и 15% пациентов в группах 1 и 2. При этом исходный уровень HBsAg коррелировал с пропорцией ответивших на лечение больных. Так, при низком изначальном уровне HBsAg (≤ 1000 МЕ/мл) первичная конечная точка была зафиксирована для 24% и 41% испытуемых, тогда как при высоком его уровне (≤ 3000 МЕ/мл) — для 14% и 26% [1].
Большинство пациентов (58% в каждой группе), показавших ответ на лечение по завершении применения бепировирсена, не столкнулись с рецидивом инфекции в ходе назначения интерфероновой терапии. Другими словами, добавление интерферона снизило риск рецидива хронического вирусного гепатита B.
Однако только 2 человека (в группе 2) с частичным ответом после бепировирсена вышли к полному ответу в процессе добавления интерферона. То есть подключение последнего к бепировирсену не привело к значимому улучшению исходов, обусловленных снижением уровня HBsAg.
Был отмечен временный рост уровня АЛТ выше утроенной верхней границы нормы. В ходе интерфероновой терапии не было отмечено ассоциации между ростом HBsAg и АЛТ.
Серьезные нежелательные явления (НЯ), связанные с лечением, были зарегистрированы только у 1 пациента (2%). Некоторые НЯ вынудили выйти из исследования 4% испытуемых (n=4): аллергический дерматит, реакции по месту введения препарата, депрессия.
B-SURE
Продолжающееся клиническое испытание B-Sure (NCT04954859) фазы II (нерандомизированное, открытое, многоцентровое, международное) поставило своей целью выяснить, как долго сохраняется SVR-статус среди ранее прошедших лечение бепировирсеном и ответивших на него пациентов с хроническим вирусным гепатитом B из других клинических испытаний этого экспериментального препарата. Наблюдательное исследование продолжается сроком максимум 33 месяца.
Положительный SVR-статус разнесен по критерию ответа на бепировирсен: полный ответ (CR) и частичный ответ (PR). Первый, отражающий функциональное излечение хронического вирусного гепатита B, предполагает снижение уровней поверхностного антигена вируса гепатита В (HBsAg) и ДНК вируса гепатита B (HBV) ниже порогов, определяемых высокоточным ПЦР-методом: соответственно 0,05 МЕ/мл и 20 МЕ/мл. Второй — уровень HBsAg < 100 МЕ/мл и уровень ДНК HBV ниже вышеуказанного порогового.
Если говорить о пациентах из B-Clear (NCT04449029), то анализ исходов B-Sure осуществляется согласно разбивке по факту терапии нуклеозидными/нуклеотидными аналогами (NA): одна группа пациентов из B-Clear получала фоновое NA-лечение, но затем, по прошествии 3 месяцев после начала участия в B-Sure, должна была прекратить, тогда как вторая группа вообще не проходила фоновую NA-терапию.
В группе фонового NA-лечения оказались 11 человек с полным ответом, из которых 9 пациентов впоследствии, согласно протоколу исследования, прекратили прием NA-препаратов. По прошествии 6 месяцев после остановки NA-терапии полный ответ сохранился у 78% (n=7/9) участников. По прошествии еще 6 месяцев (всего 12), когда 1 больной был исключен из анализа ввиду недоступности данных наблюдений за ним, полный ответ сохранился у всех оставшихся 6 человек, то есть составил 67% (n=6/9).
Ситуация с пациентами, показавшими частичный ответ к моменту включения в B-Sure, следующая. Из 29 пациентов прием NA-препаратов должным образом прекратили 23 человека. По истечении 6 месяцев частичный ответ сохранился у 22% испытуемых (n=5/23), притом что 13% (n=3/23) продемонстрировали отложенный полный ответ. После 12 месяцев частичный ответ сохранился у 13% (n=3/23), а отложенный полный ответ — у 13% (n=3/23).
В группу, не проходившую фоновое NA-лечение, попали 16 пациентов: 11 человек с полным ответом и 5 с частичным. По прошествии 15 месяцев полный ответ сохранился у 36% (n=4/11), частичный — у 20% (n=1/5).
Таким образом, лечение хронического вирусного гепатита B при помощи бепировирсена способно привести к функциональному излечению этой инфекции, что подтверждается результатами долгосрочных наблюдений за пациентами, прекратившими всякое лечение заболевания.
ЧТО ДАЛЬШЕ
На волне обнадеживающих результатов, продемонстрированных бепировирсеном в задаче излечения хронического вирусного гепатита B, «ГлаксоСмитКляйн» запустила два идентичных опорных клинических испытания, B-Well 1 (NCT05630807) и B-Well 2 (NCT05630820), фазы III (рандомизированных, двойных слепых, плацебо-контролируемых, многоцентровых, международных), которые, если завершатся успешно, лягут в основу регистрационного досье.
Среди основных требований к взрослым участникам (n=900 и n=900): стабильная терапия нуклеозидными/нуклеотидными аналогами (NA) на протяжении не менее чем 6 месяцев; уровень HBsAg в пределах 100–3000 МЕ/мл; уровень ДНК HBV < 90 МЕ/мл; уровень АЛТ не выше удвоенной верхней границы нормы.
Испытуемые, продолжающие следовать фоновой NA-терапии, вначале проходят 24-недельный курс терапии бепировирсеном или плацебо, а затем на протяжении 24 или 48 недель получают только NA-препараты.
Первичная конечная точка эффективности лечения установлена функциональным излечением хронического вирусного гепатита B, факт которого подтверждается устойчивой вирусной супрессией (SVR) на протяжении хотя бы 24-недельного периода, оставляемого без какого-либо лечения. Исследования завершатся ближе к концу 2025 года.
В клиническом исследовании B-United (NCT06537414) фазы IIb пациентам (n=280) с хроническим вирусным гепатитом B, придерживающимся стабильной NA-терапии и находящимся в SVR-статусе, вначале назначают комбинацию из даплусирана (daplusiran) и томлигисирана (tomligisiran) — по 50 и 200 мг каждые 4 недели на протяжении 24 недель, а затем проводят 24-недельный курс лечения бепировирсеном. Результаты испытания, которое должно выяснить частоту функционального излечения инфекции, будут готовы к концу 2027 года.
Комбинация из даплусирана и томлигисирана (JNJ-3989, JNJ-73763989, GSK5637608, ARO-HBV) — фиксированная доза малых интерферирующих РНК (миРНК), таргетированных на гепатоциты и индуцирующих процесс эндогенной интерференции для расщепления транскриптов РНК HBV, экспрессируемых как из ковалентно замкнутой кольцевой ДНК (кзкДНК) HBV, так и из ДНК HBV, интегрированной в геном хозяина. Это приводит к снижению уровня всех белков HBV (HBsAg, HBeAg) и его прегеномной РНК (пгРНК) [1].
Самостоятельно сочетание даплусирана и томлигисирана снижает уровень HBsAg, причем независимо от его исходной концентрации, но обеспечить функциональное излечение хронического вирусного гепатита B не в силах. Антисмысловой олигонуклеотид бепировирсен продемонстрировал свою максимальную эффективность в отношении устойчивой потери HBsAg среди пациентов с изначально относительно низким уровнем HBsAg (≤ 3000 МЕ/мл). Отсюда и родилась гипотеза, что, если снизить уровень последнего перед назначением бепировирсена, можно увеличить пропорцию пациентов, которые выйдут к статусу функционального излечения [2].
В клиническом испытании B-Focus (NCT06497504) фазы II изучается лечение пациентов (n=150) с коинфекцией хронического вирусного гепатита B и вируса иммунодефицита человека 1 (ВИЧ-1). Последний должен находиться в статусе вирусной супрессии благодаря антиретровирусной терапии (АРТ). Результаты будут собраны к середине 2027 года.
ЧТО ЕЩЕ
«ГлаксоСмитКляйн» осуществляет клиническое исследование NCT05276297 фазы II, в котором пациентам (n=184) после 12- или 24-недельной терапии хронического вирусного гепатита B бепировирсеном следует назначение экспериментальной таргетной иммунотерапии GSK3528869A.
Первичная конечная точка эффективности лечения установлена устойчивой вирусной супрессией (SVR) по прошествии 24 недель после терапии. Испытание должно завершиться к зиме 2026 года.
GSK3528869A представляет собой иммунотерапевтическую вакцину из трех компонентов:
ChAd155-hIi-HBV: лишенный возможности реплицироваться аденовирус шимпанзе группы C серотипа 155, кодирующий последовательности двух белковых антигенов HBV: усеченного ядерного антигена вируса гепатита В (HBcAg) и полноразмерного малого поверхностного антигена вируса гепатита В (S-HBsAg);
MVA-HBV: кодирующий два вышеуказанных белковых антигена HBV модифицированный осповакцинный вирус Ankara (Modified vaccinia Ankara, MVA), представляющий собой высокоаттенуированный штамм вируса осповакцины (Vaccinia virus);
HBc-HBs/AS01B-4: вышеуказанные белковые антигены HBV, подкрепленные адъювантом AS01B-4, который представляет собой липосомальное сочетание 3-О-дезацилированного 4′-монофосфорил-липида A (MPL) сальмонеллы (Salmonella minnesota) и молекулы сапонина (QS-21) из растительного экстракта квиллайи мыльной (Quillaja saponaria).
Первый компонент GSK3528869A вводится по завершении курса бепировирсеном: в 1-й день, второй — в 57-й, третий — в 113-й и 169-й.
Концептуальная идея применения терапевтических вакцин вроде GSK3528869A состоит в том, что недостаточность индукции HBV-специфического B- и T-клеточного иммунитета ответственна за отсутствие полного клиренса вируса гепатита B [1] [2] [3] [4]. Вакцина должна запускать формирование сильного вирусоспецифического иммунитета против антигенов HBV, контролирующего инфекцию путем индукции нейтрализующих антител и элиминации инфицированных гепатоцитов при участии эффекторных T-клеток [4] [5].
В начале декабря 2024 года «ГлаксоСмитКляйн» остановила клиническую проверку NCT03866187 фазы I/II иммунотерапевтической вакцины GSK3528869A, назначаемой пациентам (n=135) с хроническим вирусным гепатитом B, находящимся в SVR-статусе благодаря терапии нуклеозидными/нуклеотидными аналогами (NA). Заявлено об отсутствии должной эффективности по прошествии 24 недель после лечения [6].
Клиническое исследование NCT05330455 фазы I/II, которое должно завершиться к концу 2027 года, тестирует среди пациентов (n=132) с хроническим вирусным гепатитом B сочетание из бепировирсена и GSK3965193, низкомолекулярного ингибитора неканонической атипичной поли(А)-полимеразы 5 и 7 (PAPD5 и PAPD7). Эти ферменты нужны для стабилизации РНК HBV посредством вирусного посттранскрипционного регуляторного элемента (PRE) [7] [8] [9]. Ингибирование PAPD5 и PAPD7 приводит к подавлению вирусной репликации и синтеза вирусных белков, включая HBsAg [10] [11] [12]. В доклинических исследованиях на мышиной модели HBV продемонстрирована оправданность комбинации бепировирсена и GSK3965193 с позиции усиления снижения уровня HBsAg [13].
ЭКСПЕРТНЫЕ КОММЕНТАРИИ
Все существующие стратегии лечения хронического вирусного гепатита B преследуют цель долгосрочной супрессии (подавления) уровня ДНК вируса гепатита B (HBV). При этом весьма желательной является потеря антигена e вируса гепатита B (HBeAg) у HBeAg-положительных пациентов, поскольку она отражает наличие частичного иммунного контроля над инфекцией. В качестве дополнительной цели следует рассматривать нормализацию уровня АЛТ.
Оптимальной конечной точкой лечения выступает устойчивая потеря HBsAg, так как она указывает на глубокую супрессию репликации HBV и экспрессии вирусного белка, свидетельствуя о функциональном излечении (вирусная супрессия на протяжении не менее чем 6 месяцев) хронического вирусного гепатита B, то есть когда вирус не полностью элиминирован (устранен) из организма, но иммунная система контролирует его без каких-либо лекарственных препаратов [1] [2].
Доступные медикаментозные подходы к лечению хронического вирусного гепатита B, представленные пэгинтерфероновой терапией и назначением нуклеозидных/нуклеотидных аналогов (NA), не могут похвастаться безоговорочной эффективностью. Так, потеря HBsAg происходит весьма редко: по прошествии 6 месяцев после годичного курса лечения это наблюдается в 3–7% случаев пэгинтерфероновой терапии и 0–3% случаев терапии NA. Если применение NA продолжается долго, скажем, 5–8 лет, вероятность потери HBsAg повышается, но опять же незначительно: до 10–12% у изначально HBeAg-положительных пациентов и до менее чем 1–2% у HBeAg-отрицательных [1].
Столь скромная эффективность лечения хронического вирусного гепатита B обусловлена тем, что полная эрадикация HBV нынешними препаратами затруднена по причине сохранения в гепатоцитах как ковалентно замкнутой кольцевой ДНК (кзкДНК) HBV, так и интегрированной в их ядро ДНК HBV, являющихся транскрипционными шаблонами для возобновления репликации ДНК HBV [3] [4].
Попытки комбинированного лечения обеспечили потерю HBsAg в 14% случаев, если после минимум 48-недельной NA-терапии переключить пациентов на пэгинтерфероновую терапию [5]. Считается, что прямая противовирусная активность NA, за счет ингибирования вирусной ДНК-полимеразы (обратной транскриптазы) приводящая к вирусологической супрессии и подавлению репликации HBV, частично восстанавливает адаптивный иммунитет, тем самым способствуя улучшению иммуномодулирующего действия пэгинтерферона, проявляющегося в предотвращении образования белков HBV и деплеции (истощении) внутрипеченочного пула кзкДНК [6] [7] [8] [9]. Тем не менее подход нуждается в дополнительных уточняющих исследованиях.
Помимо функционального излечения хронического вирусного гепатита B, существует куда менее достижимая цель стерильного излечения, когда HBsAg не обнаруживается, а ДНК HBV, включая кзкДНК и интегрированную, уничтожена.
В клиническом испытании B-Clear (NCT04449029) бепировирсен продемонстрировал высокую эффективность лечения, если отталкиваться от того факта, что потеря HBsAg установлена для почти трети пациентов после относительно короткого 24-недельного курса лечения.
Вирусологический ответ был зафиксирован среди как HBеAg-отрицательных пациентов, так и придерживающихся NA-терапии HBеAg-положительных. Это свидетельствует о том, что целевая для бепировирсена терапевтическая мРНК-последовательность HBV присутствует даже тогда, когда HBsAg получен из интегрированных вирусных геномов [10].
Отмеченный рост уровня АЛТ, сопутствовавший снижению уровня HBsAg, указывает на благотворные иммунные процессы. Известно, что повышение уровня АЛТ, суррогатного маркера воспаления печени, непрямым образом отражает факт иммуноопосредованного разрушения и клиренса инфицированных гепатоцитов и является предиктором потери HBsAg [11] [12].
Нельзя сказать, что нынешние результаты клинической проверки бепировирсена оказались разочаровывающими. Да, статус функционального излечения хронического вирусного гепатита B, подтвержденный отсутствием HBsAg и ДНК HBV на протяжении 6 месяцев после завершения лечения, зафиксирован у максимум 10% и 14% пациентов — соответственно среди проходивших фоновую терапию NA и без таковой. Однако с учетом короткого курса лечения эффективность следует воспринимать с должным оптимизмом.
Поскольку бепировирсен проявил наибольшую эффективность среди пациентов с изначально низким уровнем HBsAg, в дальнейшем следует рассматривать последний как основополагающий критерий выбора подходящих больных, которые с повышенной вероятностью извлекут пользу от лечения.
Кроме того, одним из предикторов успеха является статус HBeAg. В группе 1 среди HBeAg-отрицательных участников функционально излечились 10% и 14% — соответственно среди проходивших фоновую терапию NA и без таковой. При HBeAg-положительном статусе излечение отмечено для 6% и 0%.
К счастью, наблюдается тенденция, что большинство пациентов с хроническим вирусным гепатитом B являются HBeAg-отрицательными, то есть ДНК-последовательности HBV интегрированы в геном хозяина и являются основным источником HBsAg [10] [13].
Несмотря на множество изучаемых экспериментальных подходов к лечению хронического вирусного гепатита B [14], в их отношении назревает критика: мол, большинство из них фундаментально заблуждаются [15]. Инфекция HBV характеризуется очень высокой степенью генетической пластичности (тысячи квазивидов существуют у каждого отдельного пациента) [16], что является результатом отсутствия коррекционной активности обратной транскриптазы HBV, высокой скорости обновляемости (turnover) кзкДНК и постоянного иммунного давления, оказываемого на вирус. Учитывая, что даже одиночные точечные мутации HBV отменяют способность антисмысловых олигонуклеотидов (ASO) и малых интерферирующих РНК (миРНК) к специфическому расщеплению мРНК [17], под сомнение ставится даже теоретическая состоятельность данных классов лекарственных соединений для лечения хронического вирусного гепатита B.
На вышесказанное намекает тот факт, что снижение уровня HBsAg, обеспеченное бепировирсеном, оказалось сильнее среди пациентов с изначально более низким уровнем HBsAg (≤ 3000 МЕ/мл), что противоречит заявленному механизму действия препарата: сила снижения HBsAg не должна зависеть от его исходного уровня.
В предшествовавшем клиническом испытании NCT02981602 фазы IIa наблюдалась аналогичная картина [18]. Опять же, GSK3389404, вариант бепировирсена, конъюгированный с N-ацетилгалактозамином (GalNAc) в целях более таргетной доставки в гепатоциты [19], не оказал существенного влияния на снижение уровня HBsAg [20].
«Кавигейл» (Kavigale, сипавибарт) — новый лекарственный препарат, предназначенный для доконтактной профилактики (PrEP) коронавирусной инфекции COVID-19, вызванной коронавирусом SARS-CoV-2, у взрослых и подростков (12 лет и старше, весом как минимум 40 кг) с ослабленным иммунитетом по состоянию здоровья или ввиду приема иммуносупрессивных препаратов.
ОСНОВНЫЕ ФАКТЫ
«Кавигейл» снизит риск развития симптоматического ковида, вызванного любым вариантом коронавируса, а также риск развития осложнений инфекции, требующих госпитализации.
По сути «Кавигейл» представляет собой альтернативу стандартной противоковидной вакцине. Но применяться он должен только в том случае, если человек по каким-либо причинам, связанным со здоровьем или нынешним лечением, не может привиться от коронавируса.
«Кавигейл» вводится внутримышечной инъекцией один раз в полгода.
Сипавибарт (sipavibart) — противовирусное моноклональное антитело, наделяющее пассивной иммунизацией против SARS-CoV-2 путем блокирования проникновения коронавируса в клетки: вирус теряет возможность для своего размножения, в результате становясь неинфекционным.
«Кавигейл» предложен «АстраЗенека» (AstraZeneca) взамен ушедшего в прошлое «Эвушелда» (Evusheld, тиксагевимаб + цилгавимаб), который был первым препаратом, предназначенным для противоковидной PrEP-защиты, но который весьма быстро лишился своей эффективности ввиду появления новых омикрон-вариантов SARS-CoV-2.
В середине декабря 2024 года Комитет по лекарственным препаратам для медицинского применения (CHMP) при Европейском агентстве по лекарственным средствам (EMA) отрекомендовал одобрить «Кавигейл». Решение регулятора не за горами [1].
В США и других странах сипавибарт, если будет официально разрешен, может получить иное брендовое название: например, «Нексшелд» (Nexsheld), «Нексшелда» (Nexshelda), «Райшелда» (Ryshelda).
Пемивибарт каждые три месяца — для тех, кто не может привиться против COVID-19.
ПОЧЕМУ ЭТО ВАЖНО
По данным реальной клинической практики, люди с ослабленной иммунной системой испытывают непропорционально большое бремя коронавирусной инфекции COVID-19 по сравнению с общей популяцией, гораздо чаще сталкиваясь с тяжелым течением вызванного коронавирусом SARS-CoV-2 заболевания и его неблагоприятными последствиями.
Так, согласно обсервационному популяционному исследованию INFORM, охватившему резидентов Великобритании, люди с ослабленным иммунитетом подвержены существенно большим рискам госпитализации или смерти по причине осложнений ковида: для определенных категорий эти риски вырастают в 13 и 20 раз. Несмотря на то что таких индивидуумов насчитывается лишь 3,9% населения, они вносят весомый вклад в статистику неблагополучных ковидных исходов: в 2022 году на их долю пришлось 22% случаев госпитализации, 28% случаев поступления в отделение интенсивной терапии, 24% смертельных исходов. И всё это не взирая на то, что 84% людей с ослабленным иммунитетом получили как минимум три дозы противоковидных вакцин [1].
Согласно обсервационному когортному исследованию EPOCH-US среди резидентов США, проведенному в период с апреля 2020 года по март 2022-го, люди с ослабленным иммунитетом, которых насчитывается 2,7% населения, требуют выделения более чем вчетверо больших ресурсов здравоохранения, чем представители общей популяции, при госпитализации с коронавирусной инфекцией COVID-19 [2].
КЛИНИЧЕСКАЯ ПРОВЕРКА
Клиническое исследование SUPERNOVA (NCT05648110) фазы III (рандомизированное, двойное слепое, плацебо-контролируемое и с активным контролем, многоцентровое, международное) пригласило добровольцев (n=3335) в возрасте 12 лет и старше в целях изучения эффективности и безопасности «Кавигейла» /«Нексшелда» (Kavigale / Nexsheld, сипавибарт) в сравнении с «Эвушелдом» (Evusheld, тиксагевимаб + цилгавимаб) в задаче доконтактной профилактики коронавирусной инфекции COVID-19.
Среди основных требований к участникам: отрицательный экспресс-тест на антиген COVID-19 и наличие статуса ослабленного иммунитета по такой причине, как солидная или гематологическая злокачественная опухоль, трансплантированный солидный орган или гемопоэтические стволовые клетки, прием иммуносупрессивных препаратов, лечение CAR-T-клетками или истощающая пул B-клеток терапия, гемодиализ, умеренный или тяжелый первичный иммунодефицит.
Участникам назначали внутримышечные инъекции: либо 300 мг сипавибарта (sipavibart), либо 300 мг тиксагевимаба (tixagevimab) с 300 мг цилгавимаба (cilgavimab) — дважды с интервалом в 6 месяцев между дозами.
В этот годичный период проводилась сравнительная оценка нейтрализующей активности «Кавигейла» /«Нексшелда» и «Эвушелда», а также их эффективности в предотвращении развития симптоматической коронавирусной инфекции COVID-19, вызванной любым вариантом коронавируса SARS-CoV-2 и его вариантами без мутации F456L. Собирались также данные, касающиеся случаев симптоматического ковида, заболевания в тяжелой форме, госпитализации по по причине осложнений, смертельных исходов.
Исследование, начавшееся в середине декабря 2022 года, должно было завершиться в конце марта 2023-го. Осталось дождаться публикации полных его результатов.
«АстраЗенека» также осуществила клиническое исследование NOVELLA (NCT06057064) фазы II, проверившее «Нексшелд» среди жителей России (n=116).
ЭФФЕКТИВНОСТЬ
В ходе разработки сипавибарта (sipavibart) преследовалась следующая задача: сделать это моноклональное антитело таким образом, чтобы его нейтрализующая активность оставалась максимально высокой против, во-первых, прежде доминировавших вариантов (разновидностей) коронавируса SARS-CoV-2 и, во-вторых, тех его новых вариантов, которые становятся всё более распространенными в настоящее время.
И если первая часть этой задача вполне себе реализуема, то со второй неизбежно возникают сложности, поскольку SARS-CoV-2 проходит безостановочный процесс эволюционных генетических изменений, потенциально способных снизить эффективность любого профилактического или лекарственного препарат либо вообще превратить его в неработающий.
Вот почему нельзя безоговорочно заявлять, что сипавибарту по силам обеспечить абсолютную защиту от коронавирусной инфекции COVID-19. Тем не менее некоторые достоверные предположения сделать всё же можно — даже без наличия результатов клинической проверки препарата «Кавигейл» /«Нексшелд» (Kavigale / Nexsheld, сипавибарт).
Так, согласно данным in vitro сипавибарт сохраняет нейтрализующую активность против всех исторических вариантов SASR-CoV-2, а также омикрон-подвариантов, включая JN.1, BA.2.86 и XBB.1.16. Однако он ее теряет в случае омикрон-подвариантов с мутацией F456L, таких, например, как XBB.1.5.10 / EG.5, EG.5.1, KP.1.1, LB.1, KP.3.3 [1]. Пока нет данных, касающихся нейтрализующей активности сипавибарта против набирающих силу F456L-мутантных разновидностей KP.3.1.1 и XEC.
Согласно результатам клинической проверки SUPERNOVA (NCT05648110) сипавибарт в целом снизил риск развития симптоматической коронавирусной инфекции COVID-19 на 30% относительно «Эвушелда» (Evusheld, тиксагевимаб + цилгавимаб). Если отбросить варианты коронавируса с F456L-мутацией, снижение составило относительных 35% [2].
Европейское агентство по лекарственным средствам (EMA) настоятельно не рекомендует использовать сипавибарт в тех странах и регионах, где, согласно текущей эпидемиологической обстановке, преобладают F456L-мутантные варианты SARS-CoV-2: препарат, скорее всего, не принесет никакой клинической пользы. В таком случае единственно верным решением защиты от ковида остается вакцинация — даже лиц с ослабленной иммунной системой.
Для более точного понимания потенциальной степени защитной эффективности препарата «Кавигейл» /«Нексшелд» и принятия информированного решения на предмет оправданности его профилактического назначения, следует изучить следующие наборы данных:
Мониторинг Всемирной организации здравоохранения (ВОЗ) циркулирующих в настоящее время вариантов SARS-CoV-2, вызывающих интерес (ВВИ), и вариантов SARS-CoV-2 под наблюдением (ВПН) [3].
Распространенность того или иного варианта SARS-CoV-2 в конкретном регионе или стране [4] [5] [6] [7] [8].
Базы данных, аккумулирующие сведения о снижении чувствительности вариантов SARS-CoV-2 к нейтрализующей активности моноклональных антител [9].
Релевантные научные, исследовательские и презентационные публикации [10] [11].
Инструкция по медицинскому применению препарата «Кавигейл» /«Нексшелд» (Kavigale / Nexsheld, сипавибарт), в которой должны быть указаны подробные сведения, касающиеся нейтрализации вариантов SARS-CoV-2 с информацией о снижении чувствительности и развитии резистентности.
ВОПРОС ЦЕНЫ
Цена препарата «Кавигейл» /«Нексшелд» (Kavigale / Nexsheld, сипавибарт) будет объявлена, когда он получит регуляторное одобрение.
Можно смело полагать, что стоимость сипавибарта (sipavibart) никак не будет выставлена ниже 2 тыс. долларов. Это типичная цена для моноклональных антител против коронавируса SARS-CoV-2, когда они еще эффективно работали и были повсеместно разрешены для применения [1] [2] [3] [4] [5].
Для бизнеса «АстраЗенека» запуск сипавибарта определенно выгоден, если отталкиваться от заработков ушедшего в прошлое «Эвушелда», который принес 85 млн, 2,19 млрд и 312 млн долларов — соответственно в 2021, 2022 и 2023 гг.
КАК ЭТО РАБОТАЕТ
Сипавибарт (sipavibart, AZD3152) — полностью человеческое моноклональное IgG1-антитело, таргетированное на высококонсервативный эпитоп рецептор-связывающего домена (RBD) S-белка коронавируса SARS-CoV-2.
После связывания сипавибарта с RBD блокируется взаимодействие последнего с ангиотензинпревращающим ферментом 2 (ACE2) — рецептором, который коронавирус использует для прикрепления к мембране клеток организма-хозяина с последующим проникновением в них.
Пассивная иммунизация против ковида при помощи сипавибарта, предполагающая нейтрализацию SARS-CoV-2, который теряет возможность для репликации, обеспечивает временную защиту от заражения, а если инфекция всё же дала о себе знать, то следует ожидать замедления прогрессирования заболевания и ускорения выздоровления.
Сипавибарт является производным антител, полученных из B-клеток памяти выздоровевшего пациента, вакцинированного, но всё равно столкнувшегося с заражением коронавирусной инфекцией COVID-19, вызванной омикрон-вариантом BA.1 коронавируса SARS-CoV-2.
В антительный каркас сипавибарта внедрены аминокислотные замены, призванные улучшить его фармакокинетические и биохимические свойства [1]. Так, замена YTE повысила сродство антитела к неонатальному Fc-рецептору, что привело к улучшению рециркуляции и существенному продлению периода полувыведения так, что продолжительность действия препарата увеличилась более чем втрое по сравнению с обычными неоптимизированными антителами [2] [3] [4].
Продолжительное персистирование сипавибарта в организме позволяет надеяться, что противоковидная защита сохранится на протяжении не менее чем шести месяцев после однократного введения препарата [5]. Впрочем, учитывая агрессивную мутационную природу SARS-CoV-2, сипавибарт, не исключено, придется применять чаще, если поставлена задача максимизации защиты от ковида.
Аминокислотная замена TM снизила силу связывания с FcγR и C1q, что отразилось резким ослаблением или даже отсутствием эффекторной Fc-функции, включающей антителозависимый клеточный фагоцитоз (ADCP), антителозависимую клеточную цитотоксичность (ADCC), антителозависимое осаждение комплемента (ADCD), антителозависимую активацию естественных клеток-киллеров (ADNKA) и влекущей за собой иммунопатологические проявления. Минимизирован потенциальный риск антителозависимого усиления (ADE) — явления, при котором неоптимальные вирусоспецифические антитела, напротив, способствуют, а не подавляют инфекцию или заболевание [6].
PERICULUM IN MORA: ОПАСНОСТЬ В ПРОМЕДЛЕНИИ
В начале декабря 2021 года свет увидел «Эвушелд» (Evusheld, тиксагевимаб + цилгавимаб) — комбинация из моноклональных антител авторства «АстраЗенека» (AstraZeneca), ставшая первым (и единственным) способом доконтактной профилактики коронавирусной инфекции COVID-19 для тех, кому не подходит или противопоказана вакцинация.
Появление «Эвушелда» оказался настолько значимым событием, что журнал «Тайм» (Time) включил его в список лучших изобретений 2022 года [1] [2].
Однако уже в конце января 2023-го FDA отменило регистрацию «Эвушелда»: ввиду засилья новых омикрон-вариантов коронавируса SARS-CoV-2, с которыми он справиться не в силах, подобный способ защиты от инфекции работать перестал [3].
Одна доза моноклонального коктейля AstraZeneca защитит от ковида на протяжении полугода и дольше.
Другие фармразработчики так и не смогли предложить альтернативу «Эвушелду», что оставило ни с чем десятки миллионов людей, отчаянно нуждающихся в подобной пассивной иммунизации от ковида — вместо активной иммунизации, реализуемой посредством вакцинации.
«АстраЗенека», очевидно прогнозировавшая печальную участь «Эвушелда», заблаговременно подсуетилась, в середине мая 2022 года лицензировав сипавибарт (sipavibart) и другие противоковидные моноклональные антитела у британской «Ар-кью байо» (RQ Bio) и взамен пообещав до 157 млн долларов плюс роялти от реализации готового препарата [4].
Клиническая проверка сипавибарта началась в середине декабря 2022 года [5].
«АстраЗенека» рассчитывала сделать сипавибарт доступным для широкого применения во второй половине 2023 года [6].
Для быстрого вывода «Кавигейла» / «Нексшелда» (Kavigale / Nexsheld, сипавибарт) на открытый рынок существовали абсолютно все условия, ведь препарат взял на вооружение точно такой же глубоко оптимизированный каркас антитела, на базе которого построен «Эвушелд», то есть никаких конструктивных доработок не требовалось. Фактически стояла единственная задача: оценить защитную эффективность сипавибарта в противодействии новым вариантам коронавируса SARS-CoV-2, которые перестали реагировать на «Эвушелд».
Но не сложилось.
В середине марта 2024 года «Инвивид» (Invivyd) выпустила «Пемгарда» (Pemgarda, пемивибарт) — моноклональное антитело, предназначенное для доконтактной профилактики (PrEP) коронавирусной инфекции COVID-19, вызванной коронавирусом SARS-CoV-2, у взрослых и подростков.
Пемивибарт каждые три месяца — для тех, кто не может привиться против COVID-19.
ОБРАТНАЯ СТОРОНА
В отличие от США [1] на многих территориях, включая Россию, Европу, Великобританию, Канаду, Австралию и пр., «Эвушелд» (Evusheld, тиксагевимаб + цилгавимаб) и/или другие противоковидные моноклональные антитела и их комбинации по-прежнему включены в клинические рекомендации по профилактике и/или лечению коронавирусной инфекции COVID-19 [2] [3] [4] [5] [6].
Что примечательно, сами регуляторы давно выпустили многочисленные предупреждения о недостаточной эффективности моноклональных антител в борьбе с новыми вариантами SARS-CoV-2 в эпоху омикрон-линии, но до сих пор не отменили их маркетинговые авторизации [7] [8] [9] [10]. Не исключено, отраслевые контролеры заняли выжидательную позицию: вдруг коронавирус мутирует так, что моноклональные антитела вновь смогут с ним сражаться?
Авторитетные медицинские агентства высказываются резко против любых противоковидных моноклональных антител по причине полного отсутствия у них должной эффективности: коронавирус SARS-CoV-2 слишком глубоко и сильно мутировал, чтобы эти препараты, прежде работавшие, продолжали обеспечивать необходимую противовирусную нейтрализующую активность [11] [12] [13].
Согласно обновляемой базе данных Стэнфордского университета, отслеживающей снижение чувствительности вариантов коронавируса к нейтрализующей активности моноклональных антител, таргетированных на S-белок SARS-CoV-2, ситуация удручает: препараты этого класса в целом не пригодны к войне против ковида с его нынешним набором мутаций [14].
Кроме «Пемгарда» (Pemgarda, пемивибарт) — да и то уже с большой натяжкой! — никакие из когда-либо официально одобренных противоковидных моноклональных антител, пусть даже ранее относительно успешно применявшихся в профилактике коронавирусной инфекции COVID-19 (доконтактной и постконтактной) и ее лечении, сейчас использовать нет никакого смысла. Среди таких препаратов, в настоящее время абсолютно бесполезных [15] [16] [17] [18] [19] [20] [21] [22] [23] [24]:
Позиция доказательной медицины проста и понятна: зачем платить большие деньги за абсолютно никчемные препараты и при этом тщетно обнадеживать людей, будто бы они верно защищены от ковида?
Экспериментальный препарат BNT327 (PM8002) может стать новым стандартом первоочередного лечения трижды негативного рака молочной железы.
ОСНОВНЫЕ ФАКТЫ
Препарат-кандидат BNT327 (PM8002) продемонстрировал обнадеживающие промежуточные результаты клинической проверки прежде нелеченного неоперабельного местнораспространенного или метастатического трижды негативного рака груди.
Экспериментальная терапия сработала одинаково эффективно вне зависимости от уровня опухолевой экспрессии PD-L1.
Сейчас в первоочередной терапии при данном показании применяется «Китруда» (Keytruda, пембролизумаб), блокатор PD-1 авторства «Мерк и Ко» (Merck & Co.), но он требует наличия высокого уровня PD-L1-экспрессии.
BNT327 (PM8002) — биспецифическое антитело, одновременно нацеленное на PD-L1 и VEGF-A.
BNT327 (PM8002) разработан китайской «Байотеус» (Biotheus).
В ноябре 2023 года немецкая «Байонтек» (BioNTech) лицензировала BNT327 (PM8002) у «Байотеус» для его разработки и продвижения за пределами Китая [1]. Через год, в ноябре 2024-го, «Байонтек» купила всю «Байотеус» за авансовых 800 млн долларов и последующие выплаты до 150 млн долларов, получив доступ к обширному портфелю из экспериментальных биспецифических антител [2].
Пембролизумаб проник в запретную онкологическую зону.
КАК ЭТО РАБОТАЕТ
BNT327 (PM8002) представляет собой биспецифическое антитело, содержащее две тяжелых цепи (VHH) гуманизированного антитела против PD-L1, слитых с C-концевым иммуноглобулином G (IgG) против VEGF-A.
Одновременное таргетирование на PD-L1 и VEGF-A, во-первых, блокирует взаимодействие PD-1 и PD-L1, тем самым обращая вспять иммуносупрессивный и истощенный фенотип эффекторных Т-клеток и усиливая опосредованный цитотоксическими Т-лимфоцитами иммунный ответ против опухолевых клеток, и, во-вторых, захватывает и нейтрализует выделяемый опухолевыми клетками VEGF-A, способствуя нормализации состояния кровеносных сосудов, склонных к разрастанию и питанию опухоли.
КЛИНИЧЕСКИЕ ПОДРОБНОСТИ
Продолжающееся клиническое исследование NCT05918133 фазы Ib/II пригласило взрослых пациентов (n=42) из Китая с прежде нелеченным неоперабельным местнораспространенным или метастатическим трижды негативным раком молочной железы.
Участникам назначали внутривенно PM8002 в сочетании с наб-паклитакселом — до момента неприемлемой токсичности или прогрессирования заболевания.
После наблюдений на протяжении медианных 18,1 месяца (95% ДИ [здесь и далее]: 16,9–19,7) промежуточные результаты получились следующими [1] [2]:
частота общего ответа (ORR): 79% (63–90), включая 2% (n=1/42) полных ответов (CR) и 76% (n=32/42) частичных ответов (PR);
частота подтвержденного общего ответа (cORR): 74% (58–86);
частота контроля заболевания (DCR) как сумма частот CR, PR и стабилизации заболевания (SD): 95% (84–99);
медиана времени до ответа (TTR): 1,9 месяца (1,8–2,0);
медиана длительности ответа (DOR): 11,7 месяца (7,2–17,3);
медиана выживаемости без прогрессирования (PFS): 13,5 месяца (9,4–18,1);
медиана общей выживаемости (OS): еще не созрела;
12-месячная частота OS: 81% (65–90);
15-месячная частота OS: 78% (62–88);
18-месячная частота OS: 72% (55–84).
При разбивке пациентов сообразно отсутствию (CPS < 1; n=13) или наличию (CPS ≥ 1; n=25) опухолевой экспрессии PD-L1 показатель cORR составил 77% и 72%. При высокой PD-L1-экспрессии (CPS ≥ 10) все пациенты (n=9) достигли частичного ответа.
ЧТО ДАЛЬШЕ
Продолжаются два клинических испытания лечения неоперабельного местнораспространенного или метастатического трижды негативного рака груди:
NCT06419621 фазы III: первоочередная терапия — PM8002 с наб-паклитакселом в сравнении только с последним;
NCT06449222 фазы II: терапия первой или второй линии — BNT327 в комбинации с или наб-паклитакселом, или паклитакселом, или эрибулином, или карбоплатином и гемцитабином.
ЧТО ЕЩЕ
«Байонтек» и «Байотеус» верят в успех BNT327 (PM8002), и поэтому настроены весьма серьезно, масштабно проверяя этот экспериментальный препарат в лечении различных онкологических заболеваний:
NCT06712355 фазы III: прежде нелеченный мелкоклеточный рак легкого (МРЛ) на распространенной стадии — BNT327 с химиотерапией (этопозид + карбоплатин) в сравнении с «Тецентриком» (Tecentriq, атезолизумаб) с такой же химиотерапией.
NCT05844150 фазы II/III: такое же исследование, как NCT06712355, но в Китае.
NCT06616532 фазы III: терапия второй линии МРЛ, который прогрессировал после первоочередной платиносодержащей химиотерапии, — PM8002 с паклитакселом в сравнении с топотеканом или паклитакселом.
NCT06712316 фазы II/III: ранее нелеченный неоперабельный немелкоклеточный рак легкого (НМРЛ) на стадии IIIB/C или IV — BNT327 с химиотерапией (карбоплатин + пеметрексед) в сравнении с «Китрудой» (Keytruda, пембролизумаб) с такой же химиотерапией.
NCT05756972 фазы II/III: местнораспространенный или метастатический EGFR-мутантный НМРЛ на стадии IIIB/C или IV, который прогрессировал после назначения тирозинкиназного EGFR-ингибитора, — PM8002 с химиотерапией (карбоплатин + пеметрексед) в сравнении с последней.
NCT05864105 фазы II: первоочередное лечение неоперабельной местнораспространенной или метастатической гепатоцеллюлярной карциномы — PM8002 на фоне химиотерапевтической схемы FOLFOX.
NCT06584071 фазы Ib/II: первоочередное лечение неоперабельной местнораспространенной или метастатической гепатоцеллюлярной карциномы — мононазначение PM8002 или в комбинации с PM1009, биспецифическим антителом против TIGIT и PVRIG, в сравнении с «Тецентриком» и бевацизумабом.
NCT05918107 фазы II: первоочередное лечение неоперабельной злокачественной мезотелиомы — PM8002 в сочетании с пеметрекседом, цисплатином и карбоплатином.
NCT05879055 фазы II: второлинейная терапия неоперабельной нейроэндокринной опухоли, прогрессировавшей после платиносодержащей химиотерапии, — PM8002 на фоне химиотерапевтической схемы FOLFIRI.
ЭКСПЕРТНЫЕ КОММЕНТАРИИ
Лечение первой линии местнораспространенного или метастатического трижды негативного рака молочной железы при помощи BNT327 (PM8002) в сочетании с химиотерапевтическим наб-паклитакселом продемонстрировало клинически значимые исходы по общей выживаемости и противоопухолевую активность вне зависимости от статуса опухолевой экспрессии PD-L1.
Профиль безопасности экспериментального препарата оставался в рамках известных нежелательных явлений, свойственных отдельным блокаторам PD-(L)1, ингибиторам VEGF и наб-паклитакселу.
«Китруда» одобрен в первоочередном лечении при таком же показании — в сочетании с химиотерапией. Однозначное условие: высокая опухолевая PD-L1-экспрессия (CPS ≥ 10).
В клиническом испытании KEYNOTE-355 (NCT02819518), в котором пембролизумаб был проверен в комбинации с паклитакселом, наб-паклитакселом или карбоплатином с гемцитабином, результаты таковы [1]:
cORR: 53% (46–59), включая CR 17% и PR 35%;
медиана DOR: 12,8 месяца (9,9–25,9);
медиана PFS: 9,7 месяца (7,6–11,3) — отношение риска (hazard ratio, HR) 0,66 (0,50–0,88);
медиана OS: 23,0 месяца (19,0–26,3) — HR 0,73 (0,55–0,95).
В случае просто наличия экспрессии PD-L1 (CPS ≥ 1) добавление пембролизумаба к химиотерапии не смогло статистически значимым образом превзойти последнюю.
Таким образом, получается, что BNT327 (PM8002) потенциально способен заменить собой «Китруду», поскольку работает на более широкой популяции пациентов с трижды негативным раком груди.
Вообще же BNT327 (PM8002) располагает уже доказанной применимостью при различных онкозаболеваниях, включая мелкоклеточный рак легкого (МРЛ), немелкоклеточный рак легкого (НМРЛ), рак шейки матки, платинорезистентный рак яичника, почечно-клеточная карцинома.
«Мерк и Ко» не дремлет: в середине ноября 2024 года она выдала китайской «Ланова медисинз» (LaNova Medicines) авансом 588 млн долларов и пообещав еще до 2,7 млрд долларов по мере развития проекта, взамен лицензировав биспецифическое антитело LM-299, в котором антитело против VEGF слито с двумя C-концевыми однодоменными антителами против PD-1 [2]. Сейчас LM-299 проходит клиническое исследование NCT06650566 фазы I/II среди китайских пациентов (n=230) с распространенными солидными опухолями.
В конце октября 2024 на сцену вышел биотехнологический стартап «Оттимо фарма» (Ottimo Pharma), который занимается разработкой янкистомига (jankistomig), бифункционального антитела против PD-1 и VEGFR2, намеревающегося перейти к клинической проверке в 2025 году. Уникальной особенностью молекулы является то, что она связывает VEGFR2, преимущественно экспрессирующий в микроокружении опухоли, — вместо связывания VEGF по всему организму; другими словами, лечение окажется, как ожидается, более переносимым [3].
Тогда же «Кресент байофарма» (Crescent Biopharma) сообщила об обратном слиянии с «Глайкомиметикс» (GlycoMimetics), чтобы стать публичной: первая, принадлежащая «Парагон терапьютикс» (Paragon Therapeutics), пробует силы с CR-001, тетравалентной биспецифической молекулой, сочетающей два IgG-антитела против VEGF и два одноцепочечных вариабельных фрагмента (scFv) против PD-1 и готовящейся к клиническим исследованиям в конце 2025 года — начале 2026-го [4].
Интерес к одновременной блокаде PD-(L)1 и VEGF по-настоящему захлестнул фармотрасль, когда экспериментальный ивонесцимаб (ivonescimab) существенно эффективнее, чем пембролизумаб, справился с первоочередным лечением НМРЛ, причем вне привязки к уровню опухолевой экспрессии PD-L1 или гистологии [5].
В Китае ивонесцимаб, придуманный местной «Акесо» (Akeso), уже одобрен под брендом «Идафанг» (Idafang). На западе его разработкой занимается «Саммит терапьютикс» (Summit Therapeutics).
Впервые удалось серьезным образом опередить пембролизумаб в эффективности лечения рака легкого.
Промежуточные результаты клинического исследования NCT05227664 фазы II первоочередного лечения ивонесцимабом с паклитакселом или наб-паклитакселом пациентов (n=30) с местнораспространенным или метастатическим трижды негативным раком груди по прошествии медианных 10,2 месяца наблюдений [6] [7]:
ORR: 72%;
DCR: 100%;
медиана DOR: 7,5 месяца (3,9–NE);
медиана PFS: 9,3 месяца (6,2–NE).
Согласно уровню опухолевой PD-L1-экспрессии CPS < 1 (n=16), CPS < 10 (n=24) и CPS > 10 (n=6), показатель ORR составил 87%, 70% и 83%.
Экспериментальный тавападон (tavapadon) успешно прошел три из четырех клинических испытаний фазы III симптоматического лечения болезни Паркинсона.
ОСНОВНЫЕ ФАКТЫ
«ЭббВи» (AbbVie) готовится предложить тавападон — новый пероральный препарат, предназначенный для улучшения двигательных (моторных) функции у пациентов с болезнью Паркинсона.
Тавападон работает как при раннем паркинсонизме, так и при запущенной его форме.
Тавападон может применяться монотерапевтически и поверх стандартной леводопы.
В 2025 году «ЭббВи» собирается отправить регистрационное досье тавападона в адрес Управления по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA).
GLP1R-агонисты вроде ликсисенатида, эксенатида или семаглутида сдерживают прогрессирование моторных нарушений при болезни Паркинсона.
ПРЯМАЯ РЕЧЬ
«Болезнь Паркинсона является быстрее всего распространяющимся нейродегенеративным заболеванием в мире, и существует значительная потребность в новом варианте лечения, который обеспечит правильный баланс дофаминовой сигнализации и обеспечит устойчивый двигательный контроль без обременительных побочных эффектов, связанных с существующими методами лечения».
Хьюберт Фернандес (Hubert Fernandez), ведущий исследователь мирового масштаба, директор Центра восстановления неврологических функций (Center for Neurological Restoration) Кливлендской клиники (Cleveland Clinic, Кливленд, шт. Огайо, США).
«Новаторский механизм действия тавападона, избирательно активирующего дофаминовые рецепторы D1/D5, продемонстрировал потенциал, позволяющий людям, страдающим болезнью Паркинсона, найти оптимальный баланс между двигательным контролем, безопасностью и переносимостью».
Раймонд Санчес (Raymond Sanchez), медицинский директор «Серевел терапьютикс» (Cerevel Therapeutics).
«Накапливаемые положительные данные клинических испытаний тавападона еще больше подтверждают его терапевтический потенциал для людей, страдающих болезнью Паркинсона».
Примал Каур (Primal Kaur), старший вице-президент «ЭббВи» (AbbVie) по иммунологии, неврологии, уходу за глазами и развитию специальных направлений.
КАК ЭТО РАБОТАЕТ
Дофамин — это нейромедиатор, который управляет моторными (двигательными) функциями благодаря сложному взаимодействию между полосатым телом (corpus striatum) — областью мозга, отвечающей за двигательный контроль, — таламусом и моторной корой. Пациенты с болезнью Паркинсона теряют нейроны, вырабатывающие дофамин, в области мозга, известной как компактная часть (pars compacta) черного вещества (substantia nigra), что приводит ко всё большему снижению уровня дофамина в полосатом теле. Это, как считается, и обусловливает двигательные симптомы паркинсонизма [1] [2] [3] [4].
Из двух известных семейств дофаминовых рецепторов, D1-подобных и D2-подобных, подтипы D1 и D5 экспрессируются в подмножестве нейронов, чья функция заключена в модуляции сигналов от таламуса к коре головного мозга. Этот путь известен как прямой двигательный путь и отвечает за соответствующую инициацию двигательной активности. Подтипы D2, D3 и D4, экспрессируемые другой группой нейронов, передают сигналы по непрямому моторному пути, который косвенно регулирует передачу сигналов от таламуса к коре головного мозга. Этот путь приводит к торможению двигательной активности. Баланс между этими двумя группами нейронов обеспечивает надлежащий двигательный контроль [5].
Пероральный тавападон (tavapadon, PF-06649751, CVL 751) — низкомолекулярный высокоселективный сильный частичный агонист дофаминовых рецепторов D1 и D2. Он не обладает существенным сродством функциональной активностью по отношению к D2-подобным дофаминовым рецепторам (D2, D3, D4). Молекула характеризуется смещенным агонизмом к Gs-связанной сигнализации в D1-подобных дофаминовых рецепторах [6] [7] [8].
Тавападон разработан для улучшения двигательных симптомов при болезни Паркинсона. Тавападон дифференцированно активирует прямой двигательный путь, потенциально способствуя улучшению двигательной активности и одновременно минимизируя побочные эффекты, характерные для препаратов, неселективно стимулирующих дофамин, такие как дневная седация или сонливость, нарушение контроля над импульсами и риск развития психотических симптомов, включая галлюцинации. Тавападон активирует подтипы рецепторов дофамина на уровнях, обеспечивающих максимальное моторное преимущество для пациента, при этом снижая длительное перевозбуждение и десенситизацию рецепторов, которые вызываются полными агонистами, что могло бы приводить к дискинезиям и усугублению времени в состоянии «выключено» [8] [9].
Да, такие одобренные дофаминомиметики, как прамипексол (pramipexole) и ропинирол (ropinirole), являющиеся селективными агонистами дофаминовых рецепторов D2 и D3, работают неплохо, но им сопутствуют специфические и неприятные побочные реакции в виде повышенной сонливости, галлюцинаций и расстройства привычек и влечений (расстройства побуждений) [10] [11] [12]. И потому востребованы новые лекарственные средства против паркинсонизма, эффективные и безопасные.
Не исключено, у тавападона получится стать альтернативой (или хотя бы снизить ее дозы) традиционной леводопе (levodopa), синтетического предшественника дофамина, которая ответственна за появление инвалидизирующих и непредсказуемых моторных флуктуаций после трех–пяти лет своего назначения у более чем 40% пациентов с болезнью Паркинсона [13] [14] [15] [16] [17].
Бунтанетап действует сразу на три нейротоксичных белка, ответственных за нейродегенеративные нарушения, — бета-амилоид, альфа-синуклеин и тау-белок.
КЛИНИЧЕСКИЕ ПОДРОБНОСТИ
Тавападон справился с тремя 27-недельными клиническими испытаниями, в которых он был изучен монотерапевтически и при добавлении к леводопе — среди пациентов с болезнью Паркинсона на ранней стадии и в запущенной форме соответственно.
TEMPO-1
В клиническом исследовании TEMPO-1 (NCT04201093) фазы III ежедневная монотерапия тавападоном взрослых (40–80 лет) пациентов (n=529) с болезнью Паркинсона на ранней стадии (стадия паркинсонизма 1, 1,5 или 2 по Хён и Яру, длительность заболевания не дольше 3 лет) обеспечила статистически значимое улучшение моторных функций относительно плацебо.
Комбинированный балл, согласно частей II и III унифицированной рейтинговой шкалы оценки болезни Паркинсона Международного общества изучения двигательных расстройств (MDS-UPDRS), в 5- и 15-мг дозовых группах тавападона снизился на 9,7 и 10,2 пункта — против его роста на 1,8 пункта в группе плацебо (p<0,0001) [1].
Назначение тавападона также привело к статистически и клинически значимому расхождению с плацебо в том, что касается изменения балла по части II шкалы MDS-UPDRS.
Все подробности будут раскрыты позже.
TEMPO-2
В клиническом исследовании TEMPO-2 (NCT04223193) фазы III ежедневная монотерапия тавападоном взрослых (40–80 лет) пациентов (n=304) с болезнью Паркинсона на ранней стадии (стадия паркинсонизма 1, 1,5 или 2 по Хён и Яру, длительность заболевания не дольше 3 лет) отразилась улучшением моторных функций.
Особенностью испытания является гибкое дозирование тавападона: 5–15 мг.
Комбинированный балл, согласно частей II и III шкалы MDS-UPDRS, изменился статистически значимым образом относительно плацебо: −10,3 пункта — против −1,2 пункта (p<0,0001) [1].
В группе тавападона также зарегистрировано статистически и клинически значимое расхождение с плацебо, если говорить об изменении балла по части II шкалы MDS-UPDRS.
Все подробности будут раскрыты позже.
TEMPO-3
В клиническом исследовании TEMPO-3 (NCT04542499) фазы III ежедневное добавление тавападона (5–15 мг) к леводопе, проверенное среди взрослых (40–80 лет) пациентов (n=507) с запущенной болезнью Паркинсона (стадия паркинсонизма 2, 2,5 или 3 по Хён и Яру в состоянии «включено», наличие моторных флуктуаций), привело к продлению общего времени в состоянии «включено» без беспокоящей дискинезии на 1,7 часа — против его продления на 0,6 часа при назначении только леводопы: разница 1,1 часа (p<0,0001) [1].
Применение комбинации из тавападона и леводопы также отразилось статистически значимым сокращением времени в состоянии «выключено».
Все подробности будут раскрыты позже.
ЧТО ДАЛЬШЕ
Продолжается открытое 58-недельное клиническое исследование TEMPO-4 (NCT04760769), оценивающее долгосрочные безопасность, переносимость и эффективность гибкого дозирования тавападона среди взрослых (40–80 лет) участников (n=992) предыдущих испытаний. Исследование должно завершиться к началу 2026 года.
За разработкой тавападона стоит «Серевел терапьютикс» (Cerevel Therapeutics) — совместное предприятие, сформированное в октябре 2018 года «Пфайзер» (Pfizer) и частным инвестиционным фондом Bain Capital [1], после того как американский фармацевтический гигант решил самостоятельно не заниматься нейронауками.
В начале августа 2024 года «ЭббВи» купила «Серевел» за 8,7 млрд долларов наличными [2] [3].
Маридебарт кафраглутид (maridebart cafraglutide), разрабатываемый «Амджен» (Amgen) для долгосрочного лечения ожирения, обеспечил снижение веса на 20% за 52 недели терапии.
ОСНОВНЫЕ ФАКТЫ
Маридебарт кафраглутид, также известный как «Маритид» (Maritide), представляет собой агонист глюкагоноподобного пептида-1 (GLP-1) и антагонист глюкозозависимого инсулинотропного полипептида (GIP).
Подобное сочетание механизмов действия выглядит уникальным: пресловутый тирзепатид (tirzepatide), продвигаемый «Илай Лилли» (Eli Lilly) как «Мунджаро» (Mounjaro) для терапии сахарного диабета 2-го типа и как «Зепбаунд» (Zepbound) для лечения ожирения, является, напротив, двойным агонистом GLP-1 и GIP.
«Маритид» назначается подкожными инъекциями очень редко: один раз в месяц — тирзепатид требует введения один раз в неделю. Не исключено, в режиме поддержания веса при достигнутой цели похудения маридебарт кафраглутид можно будет применять еще реже: один раз в два месяца или даже в квартал.
«Маритид» характеризуется приемлемой переносимостью: похоже, его использовать сопровождается менее выраженными нежелательными явлениями со стороны желудочно-кишечного тракта.
«Амджен» готовится к запуску регистрационной клинической программы фазы III.
КАК ЭТО РАБОТАЕТ
Глюкагоноподобный пептид-1 (GLP-1) и глюкозозависимый инсулинотропный полипептид (GIP) — инкретиновые гормоны, вырабатываемые в кишечнике, которые усиливают стимулированную глюкозой секрецию инсулина и которые , играют важную роль в регуляции веса. GLP-1, помимо инкретиновых эффектов, способствует насыщению [1], а GIP — накоплению липидов и регуляции метаболизма [2] [3] [4] [5].
Несколько агонистов рецептора GLP-1 (GLP1RA) одобрены для лечения сахарного диабета 2-го типа, а также в целях долгосрочной коррекции избыточной массы тела при ожирении [6] [7].
Фармотрасль продолжает разрабатывать комбинированные подходы, поскольку считается, что задействование дополнительных сигнальных путей приведет к большему снижению веса, устойчивому характеру похудения и улучшению переносимости [8]. Перспективными видятся одномолекулярные решения в виде мультиспецифических пептидных агонистов, нацеленных на несколько инкретиновых путей и не только: помимо GLP-1 и GIP таргетирование осуществляется на глюкагон (GCG), амилин, пептид YY (PYY), нейропептид Y (NPY) [8] [9] [10] [11] [12] [13] [14] [15] [16] [17] [18] [19] [20].
Действительно, аналоги GIP снижают вес [21], а двойные агонисты рецептора GIP (GIPR) и рецептора GLP-1 (GLP1R) — усиливают инкретиновый ответ и снижение массы тела по сравнению с агонистами только GLP1R [22] [23] [24].
Тем не менее известно, что GIP способствует адипогенезу in vivo и ex vivo [25] [26], а уровень циркулирующего GIP повышен у мышей и людей с ожирением [27] [28] [29], что указывает на прообезогенное состояние, ассоциированное с GIP.
Более того, согласно полногеномным исследованиям ассоциаций (GWAS) у людей, локус GIPR вносит вклад в регуляцию массы тела, тогда как мыши с нокаутом GIPR защищены от алиментарно-индуцированного ожирения (DIO) [30] [31] [32] [33] [34] [35].
Фармакологическое ингибирование GIPR при помощи анти-GIPR-антител предотвратило рост массы тела у мышей с ДИО и обезьян с ожирением [36]. Антагонизм GIPR в сочетании с агонизмом GLP1R синергично снизил массу тела у тех же животных, что позволило предположить потенциал биспецифической молекулы, комбинирующей антагонизм GIPR с агонизмом GLP1R. Так и произошло: серия антител-антагонистов GIPR, конъюгированных с пептидными аналогами GLP-1, продемонстрировала усиленное снижение веса и улучшение метаболических параметров, если сравнивать с агонистами GLP1R [37].
Маридебарт кафраглутид (maridebart cafraglutide, AMG 133), также известный как «Маритид» (MariTide), — биспецифический конъюгат, составленный из полностью человеческого моноклонального антитела против GIPR и пептидного аналога GLP-1 (агониста GLP1R), связанных между собой аминокислотными линкерами [38].
Уникальной особенностью маридебарта кафраглутида является ингибирование GIPR. Для понимания: тирзепатид (tirzepatide), предлагаемый «Илай Лилли» (Eli Lilly) под брендами «Мунджаро» (Mounjaro) и «Зепбаунд» (Zepbound), напротив, является двойным агонистом GIPR и GLP1R.
КЛИНИЧЕСКИЕ ПОДРОБНОСТИ
Клиническое исследование NCT05669599 фазы II пригласило взрослых человек (n=592) либо с лишним весом (индекс массы тела [ИМТ] ≥ 27 кг/м2) и сопутствующим ему патологическим состоянием (гипертония, дислипидемия, обструктивное апное во сне, сердечно-сосудистое заболевание), либо с ожирением (ИМТ ≥ 30 кг/м2).
Когорта A охватила участников (n=465) без сахарного диабета 2-го типа (СД2), в когорту B попали пациенты (n=127) с СД2.
На протяжении 52 недель испытуемые из когорты A получали подкожные инъекции либо ежемесячно плацебо, либо маридебарта кафраглутида по одной из следующих схем: или ежемесячно по 140, 280 или 420 мг, или один раз в два месяца по 420 мг, или ежемесячно по 420 мг после 4- и 12-недельного постепенного повышения дозы.
В течение такого же периода времени пациентам из когорты B назначали ежемесячно либо плацебо, либо маридебарт кафраглутид по 140, 280 или 420 мг.
Итоговое уменьшение массы тела составило максимум 20% и 17% в когорте A и когорте B соответственно. В популяции диабетиков отмечено снижение уровня гликированного гемоглобина (HbA1c) на 2,2% и уровня глюкозы в крови на 58 мг/дл [1].
Примечательно, что плато сброса веса не наблюдалось, то есть продолжение лечения предполагает дальнейшее похудение.
Среди прочих благотворных терапевтических эффектов «Маритида»: снижение систолического артериального давления, уровня холестерина липопротеинов низкой плотности (ЛПНП), триглицеридов и высокочувствительного C-реактивного белка.
Применение маридебарта кафраглутида характеризовалось приемлемой переносимостью. Среди наиболее распространенных нежелательных явлений (НЯ): таковые со стороны желудочно-кишечного тракта, включая тошноту и рвоту. НЯ носили в основном легкую степень выраженности, были преходящими, эпизодическими.
Попутно «Амджен» развеяла витавшие в отрасли сомнения в безопасности «Маритида»: мол, препарат, обеспечивающий быструю потерю веса, приводит к снижению минеральной плотности костей. Предприятие поспешило заверить, что не видит связи между приемом маридебарта кафраглутида и изменением этого показателя [2].
Все клинические данные будут раскрыты позже.
ЧТО ДАЛЬШЕ
Участникам NCT05669599, которые похудели хотя бы на 15%, было предложено продолжить клиническую проверку маридебарта кафраглутида в целях дальнейшего снижения веса или поддержания достигнутой массы тела. Испытуемые получают либо плацебо, либо «Маритид» по одной из следующей схем: или ежемесячно по 70 мг, 140 или 420 мг, или один раз в квартал по 420 мг.
«Амджен» готовится к запуску клинической программы MARITIME фазы III, результаты которой лягут в основу регистрационного досье маридебарта кафраглутида для лечения ожирения и ассоциированных с ним состояний.
ЧТО ЕЩЕ
«Маритид» проходит параллельную клиническую проверку NCT06660173 фазы II в терапии пациентов с СД2.
ТЕМ ВРЕМЕНЕМ
В начале декабря 2024 года датский биотехнологический стартап «Антаг терапьютикс» (Antag Therapeutics) привлек 80 млн евро, которые пойдут на клиническую проверку экспериментального пептида AT-7687 — антагониста GIPR, назначаемого еженедельными подкожными инъекциями для лечения ожирения [1].
Идея состоит в том, чтобы AT-7687 применялся с уже существующими или еще разрабатываемыми препаратами против ожирения, включая агонисты GLP1R, — в целях решения вопросов, связанных с переносимостью, потерей мышечной массы, недостаточной степенью похудения. Новинка — в отличие от маридебарта кафраглутида авторства «Амджен», комбинирующего антагонизм GIPR с агонизмом GLP1R в одной молекуле, — обеспечит гибкость оптимального дозирования: нужные дозы как AT-7687, так и агониста GLP1R будут подбираться отдельно, с учетом специфики конкретного пациента и поставленных задач. Кроме того, AT-7687 можно будет использовать в качестве единственного препарата для поддержания сброшенного веса.
ЭКСПЕРТНЫЕ КОММЕНТАРИИ
Несмотря на привлекательность применения «Маритида» один раз в месяц, отраслевые эксперты выказали глубокое разочарование, повлекшее за собой падение биржевых котировок «Амджен».
И действительно, если вспомнить, что в клиническом испытании NCT04478708 фазы I ежемесячная 420-мг доза маридебарта кафраглутида избавила от 14,5% лишнего веса за 12 недель, то 52-недельная эффективность, выразившаяся в похудении на 20%, не выглядит особо впечатляющей [1].
Однако не всё настолько пессимистично.
Во-первых, отсутствие плато в ходе снижение веса открывает возможности для более устойчивого эффекта при долгосрочном поддержании сброшенной массы тела.
Во-вторых, для указанной задачи «Маритид» может применяться очень редко: к примеру, не чаще одного раза в два месяца или даже в квартал.
В-третьих, длительный период полувыведения маридебарта кафраглутида предоставляет продолжительную терапевтическую эффективность, но не связан с такими же длительными по времени нежелательными явлениями вроде тошноты и рвоты, как в случае «обычных» GLP1RA-препаратов, которые назначают один раз в неделю.
Во всём мире рак мочевого пузыря занимает десятое место среди наиболее часто диагностируемых видов онкологических заболеваний.
В развитых странах рак мочевого пузыря в подавляющем большинстве случаев характеризуется уротелиальной гистологией (ранее относился к переходно-клеточному раку).
Уротелиальная карцинома мочевого пузыря в три четверти случаев классифицируется как немышечно-инвазивный рак мочевого пузыря (НМИРМП), то есть как рак мочевого пузыря без прорастания в мышечный слой.
После трансуретральной резекции (ТУР) всей видимой опухоли, как первоочередного хирургического лечения НМИРМП, с последующей поддерживающей иммунотерапией бациллой Кальметта — Герена (БЦЖ), которая весьма эффективна, многие пациенты всё равно сталкиваются с рецидивом или прогрессированием заболевания.
Существует необходимость в новых способах лечения, дополняющих ТУР и БЦЖ-вакцину и преследующих цель сдерживания заболевания.
ЧТО ПРОИЗОШЛО
«Си-джи онколоджи» (CG Oncology) разработала кретостимоген гренаденорепвек (cretostimogene grenadenorepvec) — онколитический вирус, предназначенный для лечения высокорискового немышечно-инвазивного рака мочевого пузыря (НМИРМП), который не ответил на стандартную иммунотерапию бациллой Кальметта — Герена (БЦЖ).
Речь идет о лечении, направленном на то, чтобы избавить пациентов от проведения оперативного вмешательства, предполагающего радикальную цистэктомию (хирургическое удаление мочевого пузыря), после которой качество жизни резко ухудшается. Ну или максимально отсрочить эту процедуру.
Кретостимоген гренаденорепвек, который безусловно будет одобрен регуляторами, получит одно из брендовых названий: «Трувезик» (Truvesic) или «Арамира» (Aramira).
ПОЧЕМУ ЭТО ВАЖНО
В три четверти случаев рака мочевого пузыря речь идет о немышечно-инвазивном раке мочевого пузыря (НМИРМП). Первоочередное лечение предполагает трансуретральную резекцию всей видимой опухоли. В целях профилактики рецидива заболевания применяют иммунотерапию бациллой Кальметта — Герена (БЦЖ). В ряде случаев НМИРМП возвращается, и варианты его дальнейшего лечения ограничены, притом что радикальная цистэктомия несет за собой резкое ухудшение качества жизни.
Немышечно-инвазивный рак мочевого пузыря (НМИРМП): варианты лечения в обозримом будущем.
КАК ЭТО РАБОТАЕТ
Кретостимоген гренаденорепвек (cretostimogene grenadenorepvec, CG0070) — онколитический вирус, избирательно реплицирующийся в опухолевых клетках с дефектом сигнального пути белка ретинобластомы (RB). Подобная избирательность реализована благодаря тому, что в последовательность аденовируса серотипа 5 (Ad5) вставлен промотор E2F. Дополнительно присутствует трансген, кодирующий гранулоцитарно-макрофагальный колониестимулирующий фактор (GM-CSF) [1].
Белок RB, будучи опухолевым супрессором, предотвращает чрезмерный рост клеток путем ингибирования прохождения по клеточному циклу, до тех пор пока клетка не будет готова к делению. Когда клетка готова к нему, RB фосфорилируется, тем самым инактивируясь, и клеточному циклу разрешается идти дальше [2]. Сверхэкспрессия RB ассоциирована с повышенным риском прогрессирования и сокращенной выживаемостью [3]. При раке мочевого пузыря его опухолевые клетки почти всегда несут RB-мутации [4].
Кретостимоген гренаденорепвек наделен двойным механизмом действия: прямым лизисом опухоли за счет селективной репликации в опухолевых клетках с дефектами сигнального пути RB и иммуноопосредованным их уничтожением в результате иммуногенной гибели опухолевых клеток и локальной выработки GM-CSF — цитокина, критически важного для иммунной активации.
После того как кретостимоген гренаденорепвек инфицировал опухолевые клетки, они начинают погибать, в результате чего высвобождаются неоантигены. GM-CSF запускает дифференцировку антигенпрезентирующих клеток (APC), захватывающих неоантигены, и сдерживает супрессорные клетки миелоидного происхождения (MDSC). Распознавание антигенов на дендритных клетках результирует активацией T-хелперов и цитотоксических лимфоцитов (CTL) наряду с иммунной активацией опухолевого микроокружения (TME) [5].
ЧТО ВЫЯСНИЛОСЬ
«Си-джи онколоджи» (CG Oncology) провела кретостимоген гренаденорепвек (cretostimogene grenadenorepvec) через ряд клинических испытаний, надежно доказавших терапевтическую состоятельность препарата «Трувезик» (Truvesic) / «Арамира» (Aramira).
BOND-003
Продолжающееся клиническое исследование BOND-003 (NCT04452591) фазы III (нерандомизированное, открытое, многоцентровое, международное) пригласило взрослых пациентов (n=112) с высокорисковым раком мочевого пузыря без прорастания в мышечный слой (немышечно-инвазивный), который не отреагировал на ранее проведенную иммунотерапию бациллой Кальметта — Герена (БЦЖ), то есть заболевание персистировало или рецидивировало в течение 12 месяцев по завершении последней.
Среди основных критериев включения в испытание:
предшествовавшая БЦЖ-терапия предусматривала получение как минимум 5–6 доз первоначального индукционного курса вместе с либо хотя бы 2–3 дозами поддерживающей БЦЖ-терапии, либо хотя бы 2–3 дозами повторного индукционного курса;
либо только персистирующая или рецидивирующая карцинома in situ (CIS), либо она же с рецидивирующей опухолью Ta или T1 (неинвазивное папиллярное новообразование или с инвазией субэпителиальной соединительной ткани) — должно развиться в течение 12 месяцев после завершения БЦЖ-терапии;
участники должны были пройти трансуретральную резекцию всех опухолей мочевого пузыря (компоненты Ta и T1);
непригодность к радикальной цистэктомии или отказ от нее.
Пациентам интравезикально назначали кретостимоген гренаденорепвек (cretostimogene grenadenorepvec) — еженедельно на протяжении первых 6 недель лечения. Если на 13-й недели высокозлокачественное заболевание сохранялось, 6-дозовый цикл повторяли. При отсутствии признаков заболевания (например, полный ответ) переходили к курсу из 3 еженедельных доз. Далее, начиная с 25-й недели, лечение осуществлялось 3 еженедельными дозами каждые 12 недель — до 49-й недели, а затем каждые 24 недели.
Включенные в исследование пациенты с медианой возраста 74 года (43–90) характеризовались весьма запущенным заболеванием: медианное число предшествовавших инстилляций БЦЖ составило 12 (7–66); некоторые также прошли терапию «Китрудой» (Keytruda, пембролизумаб), блокатором PD-1 авторства «Мерк и Ко» (Merck & Co.).
Первичная конечная точка эффективности лечения была установлена пропорцией пациентов, в любое время терапии показавших полный ответ (CR) со стороны CIS (с сопутствующим высокозлокачественным папиллярным новообразованием Ta/T1 или без него).
Согласно предварительным данным, собранным на начало октября 2023 года среди 66 пациентов, за которыми наблюдали в течение не менее чем 3 месяцев, лечение кретостимогеном гренаденорепвеком вывело к CR-статусу 76% больных (95% ДИ [здесь и далее]: 63–85; n=50/66) [1].
3-и 6-месячная частоты CR составили 68% (55–79; n=45/66) и 64% (51–75; n=42/66).
Длительность ответа (DOR) на протяжении как минимум 3 и 6 месяцев оказалась справедливой для 84% (70–92; n=42/50) и 74% (58–86; n=32/43) испытуемых.
Применение кретостимогена гренаденорепвека характеризовалось приемлемой переносимостью. Среди наиболее распространенных нежелательных явлений (НЯ): симптомы со стороны мочеполовой системы, преходящие и носившие легко-умеренную выраженность, такие как спазм мочевого пузыря (у 21% пациентов), поллакиурия (16%), дизурия (14%), императивные позывы к мочеиспусканию (12%), гематурия (11%).
Согласно обновленным на начало апреля 2024 года данным, показатель CR составил 75% (65–83; n=79/105) [2].
Назначение кретостимогена гренаденорепвека отметилось стойкостью терапевтического ответа в течение продолжительного времени. Так, 54% пациентов, прошедших повторный индукционный курс, перешли в CR-статус.
Ответ длительностью 12 месяцев и дольше показали 83% (n=29/35) пациентов. Медиана DOR не достигнута.
В период наблюдений на протяжении 1 года 92% пациентов не нуждались в радикальной цистэктомии. Ни одному из испытуемых в CR-статусе не потребовалась радикальная цистэктомия: у них не обнаруживалось ни узлового, ни метастатического прогрессирования.
12-месячная выживаемость без прогрессирования (PFS) составила 97%. Медиана PFS не зафиксирована.
Спектр наиболее распространенных НЯ легко-умеренной тяжести следующий: спазм мочевого пузыря (у 23% пациентов), поллакиурия (20%), дизурия (15%), императивные позывы к мочеиспусканию (15%), гематурия (14%).
Согласно обновленным на конец сентября 2024 года данным, лечение кретостимогеном гренаденорепвеком CR-статус сохранился у 75% пациентов (65–92; n=82/110) [3].
Медиана DOR всё еще не достигнута (как минимум, 14,5 месяца), притом что длительность ответа уже превысила 27 месяцев.
Терапевтическая эффективность впечатляет: вероятность ответа на протяжении 12 месяцев и дольше составила 64% (51–73), на протяжении 24 месяцев и дольше — 57% (43–68), а 12-месячная PFS вышла к 97%, притом что 90% испытуемых не нуждались в цистэктомии в течение этого периода.
Профиль безопасности остался прежним: спазм мочевого пузыря (у 25% пациентов), поллакиурия (21%), императивные позывы к мочеиспусканию (20%), дизурия (15%), гематурия (13%).
CORE-001
Клиническое исследование CORE-001 (NCT04387461) фазы II (нерандомизированное, открытое, многоцентровое, международное) включило взрослых пациентов с высокорисковым раком мочевого пузыря без прорастания в мышечный слой (немышечно-инвазивный), который не отреагировал на предшествовавшую иммунотерапию бациллой Кальметта — Герена (БЦЖ).
Согласно промежуточным данным, собранным на начало марта 2023 года среди пациентов (n=34), наблюдения за которым велись не менее 3 месяцев, комбинированное лечение привело к полному ответу (CR) у 85% участников (n=29/34) [1].
Статус CR на протяжении 6, 9 и 12 месяцев сохранялся соответственно у 82% (n=27/33), 81% (n=25/31) и 68% респондентов (n=17/25).
Согласно итоговым данным (n=35), собранным на начало февраля и середину мая 2024 года, по прошествии 12 месяцев в CR-статусе находились 57% пациентов (n=20/35; 40–73). Наблюдения на протяжении медианных 26,5 месяца не установили медиану длительности ответа (DOR): она превысила 21 месяц. 24-месячная частота CR вышла к 54% (n=19/35; 37–71) [2] [3].
Из тех пациентов, кто достиг статуса CR после 12 месяцев, 95% (n=19/20) сохранили этот статус сроком на дополнительных 12 месяцев.
Оценочно, вероятность выйти к полному ответу в 12- и 24-месячный периоды оказалась равной 77% (58–89) и 70% (49–83).
Никто из испытуемых не прогрессировал до мышечно-инвазивного рака мочевого пузыря или метастатического заболевания: 24-месячная выживаемость без прогрессирования (PFS) составила 100%.
Выживаемость без необходимости в цистэктомии по прошествии 24 месяцев — 80%; она же составила 100% среди пациентов с полным ответом в этот период времени.
ПЕРСПЕКТИВЫ
Будущее онколитического вируса «Трувезик» / «Арамира» (Truvesic / Aramira, кретостимоген гренаденорепвек) рисуется оптимистичным. Достаточно сравнить его клинические результаты с терапевтической эффективностью прямых конкурентов, уже одобренных для лечения немышечно-инвазивного рака мочевого пузыря (НМИРМП) после провала иммунотерапии бациллой Кальметта — Герена (БЦЖ).
Так, если монотерапия «Трувезиком» / «Арамирой» сгенерировала полный ответ (CR) у 75% пациентов, то комбинация из «Анктивы» (Anktiva, ногапендекин альфа инбакицепт) и БЦЖ выдала CR на уровне 62%, моноприменение «Адстиладрина» (Adstiladrin, надофараген фираденовек) — 51%, мононазначение «Китруды» (Keytruda, пембролизумаб) — 41%.
Кретостимоген гренаденорепвек (cretostimogene grenadenorepvec) обеспечил длительность ответа (DOR) на протяжении 12 месяцев и дольше у 83% пациентов, тогда как ногапендекин альфа инбакицепт (nogapendekin alfa inbakicept) в сочетании с БЦЖ — у 58%, а надофараген фираденовек (nadofaragene firadenovec) или пембролизумаб (pembrolizumab) — у 46%.
Более того, добавление пембролизумаба к «Трувезику» / «Арамире» оказалось успешным: 12- и 24-месячные вероятностные частоты CR составили 77% и 70%, а DOR превысила 21 месяц.
Профиль безопасности кретостимогена гренаденорепвека не вызвал каких-либо претензий: не зарегистрировано тяжелых или жизнеугрожающих нежелательных явлений.
Надофараген фираденовек позволит избежать радикальной цистэктомии.
ЧТО ДАЛЬШЕ
«Си-джи онколоджи» (CG Oncology) заинтересована в расширении популяции пригодных к назначению онколитического вируса «Трувезик» / «Арамира» (Truvesic / Aramira, кретостимоген гренаденорепвек) пациентов, и потому в конце февраля 2024 года приступила к клиническому испытания PIVOT-006 (NCT06111235) фазы III, которое проверяет препарат в лечении немышечно-инвазивного рака мочевого пузыря (НМИРМП) промежуточного риска — после трансуретральной резекции [1].
В середине сентября 2024 года было запущено клиническое исследование CORE-008 (NCT06567743) фазы II, изучающее кретостимоген гренаденорепвек (cretostimogene grenadenorepvec) при НМИРМП высокого риска среди пациентов, которые либо ранее не проходили иммунотерапию бациллой Кальметта — Герена (БЦЖ), либо прошли ее и заинтересованы в поддерживающей терапии.
Продолжается клиническое испытание NCT04610671 фазы I, тестирующее добавление «Опдиво» (Opdivo, ниволумаб), блокатора PD-1 авторства «Бристол-Майерс Сквибб» (Bristol-Myers Squibb), к «Трувезику» / «Арамире» при мышечно-инвазивном раке мочевого пузыря (МИРМП) в случае непригодности к назначению цисплатина.
БИЗНЕС
«Си-джи онколоджи» (CG Oncology), основанная в 2010 году, собрала инвестиционных денег в размере 318 млн долларов [1].
В начале января 2024 года, будучи на волне положительной позднестадийной проверки экспериментального онколитического вируса кретостимоген гренаденорепвек (cretostimogene grenadenorepvec), «Си-джи» запустила подготовку к процедуре первичного размещения на фондовом рынке (IPO) [2].
«Си-джи» собиралась привлечь сумму в диапазоне 181–209 млн долларов. Добавление имеющихся денежных средств, их эквивалентов и ценных бумаг наделило бы компанию капиталом в размере почти 370 млн долларов. Из них приблизительно 155 млн долларов планировалось пустить на финансирование исследований и разработок [3].
В конце января 2024 года «Си-джи» стала публичной компанией: в ходе IPO удалось реализовать акций на сумму 437 млн долларов [4].
На начало мая 2024 года рыночная стоимость «Си-джи» составляла 2,42 млрд долларов [5].
За оригинальной разработкой кретостимогена гренаденорепвека стоит «Селл дженисис» (Cell Genesys) [6], которая в октябре 2009 году объединилась с «Байосанте фармасьютикалс» (BioSante Pharmaceuticals) [7]. В ноябре 2010 года «Байосанте» продала «Си-джи», тогда называвшейся «Колд дженисис» (Cold Genesys), права на экспериментальное лекарство [8]. В июне 2013 года «Байосанте» провела слияние с «Эй-эн-ай фармасьютикалс» (ANI Pharmaceuticals) [9]. В марте 2024 года «Эй-эн-ай» подала на «Си-джи» в суд с требованием выплачивать роялти от реализации готового препарата [10].
ТЕМ ВРЕМЕНЕМ
Фармотрасль продолжает разрабатывать новые способы лечения немышечно-инвазивного рака мочевого пузыря (НМИРМП) высокого риска, на который не подействовала иммунотерапия бациллой Кальметта — Герена (БЦЖ). Цель прозрачна: предложить консервативное лечение, которое максимально длительно откладывает необходимость в радикальной цистэктомии.
«Атиа фармасьютикалс» (Atea Pharmaceuticals) разработала новую лекарственную комбинацию, которая позволяет навсегда вылечить хронический вирусный гепатит C (ВГС) за 8 недель.
ОСНОВНЫЕ ФАКТЫ
Сочетание бемнифосбувира (bemnifosbuvir) и рузасвира (ruzasvir) успешно прошло среднестадийную клиническую проверку.
Схема лечения предполагает ежедневный пероральный прием двух препаратов на протяжении 8 или 12 недель — соответственно без цирроза печени и с циррозом.
Бемнифосбувир с рузасвиром эффективно срабатывают при инфекции ВГС любого генотипа (включая трудноизлечимый генотип 3), а также при наличии компенсированного цирроза печени (стадия фиброза F4).
Комбинация характеризуется более чем приемлемым профилем безопасности и переносимости и отсутствием взаимодействия с другими лекарствами.
В 2025 году начнется регистрационная клиническая программа фазы III.
БОЛЬШИЕ ЧИСЛА
Несмотря на доступность противовирусных препаратов прямого действия (ПППД), представляющих собой пероральные лекарственные комбинации, хронической инфекцией вирусного гепатита C (ВГС) страдают приблизительно 50 млн человек во всём мире [1].
Ежегодно случается 1 млн новых заражений ВГС. Среди основных способов инфицирования: небезопасные инъекции, небезопасное медицинское обслуживания, переливание непроверенной крови, употребление инъекционных наркотиков, сексуальные контакты — все пути передачи обусловлены контактом с зараженной кровью.
В 2022 году хроническая инфекция ВГС повлекла за собой 242 тыс. смертельных исходов: главным образом ввиду цирроза печени и гепатоцеллюлярной карциномы (первичного рака печени).
КАК ЭТО РАБОТАЕТ
Бемнифосбувир (bemnifosbuvir, AT-527, RO7496998), будучи перорально биодоступной гемисульфатной солью AT-511, пролекарства аналога гуанозинмонофосфата, в организме в несколько этапов метаболизируется до активного 5’-трифосфата AT-9010, который действует как ингибитор РНК-полимеразы неструктурного белка 5B (NS5B), подавляющий вирусную репликацию [1] [2].
In vitro бемнифосбувир в 10 раз более активен чем софосбувир (sofosbuvir) против лабораторных штаммов и клинических изолятов вируса гепатита C генотипов 1–5. Бемнифосбувир остается полностью активным в случае мутационных замен, связанных с резистентностью к софосбувиру (S282T), при этом его эффективность всё еще в 58 раз выше, чем у последнего. Бемнифосбувир характеризуется низким риском лекарственных взаимодействий.
Рузасвир (ruzasvir, AT-038, MK-8408) — ингибитор неструктурного белка 5A (NS5A), открытый «Мерк и Ко» (Merck & Co.), которая в декабре 2021 года лицензировала его «Атиа» [3].
КЛИНИЧЕСКИЕ ПОДРОБНОСТИ
Клиническое исследование NCT05904470 фазы II проверило эффективность и безопасность ежедневно назначаемой пероральной комбинации из 550 мг бемнифосбувира и 180 мг рузасвира в лечении хронического вирусного гепатита C (ВГС) среди взрослых пациентов (n=275) без цирроза печени (стадия фиброза F0–F3) или с компенсированным циррозом (F4).
После 8 недель лечения к первичной конечной точке эффективности, установленной устойчивой вирусной супрессией, подтвержденной по прошествии 12 недель после терапии (SVR12), вышли 98% испытуемых (n=208/213), строго придерживавшихся режима приема препаратов [1].
Даже при учете тех больных, которые по каким-либо причинам пропускали очередную дозу лекарств (таковых было 17%), эффективность излечения всё равно оставалась высокой: 95% (n=242/256).
Среди комплаентных пациентов с ВГС генотипа 1–4 и без цирроза печени показатель SVR12 оказался справедливым для 99% человек (n=178/179), с циррозом — 88% (n=30/34).
Применение бемнифосбувира с рузасвиром характеризовалось приемлемой переносимостью. Нежелательные явления (НЯ) носили в целом легко-умеренную степень выраженности. Не зарегистрировано НЯ, которые либо повлекли за собой прекращение лечения, либо привели к изменению лабораторных показателей.
Дополнительные клинические данные будут представлены позже.
ЧТО ДАЛЬШЕ
«Атиа» планирует к запуску опорную клиническую проверку фазы III, которая будет представлена двумя испытания с группой активного сравнения и которая охватит большее число пациентов.
При отсутствии цирроза печени лечение будет осуществляться на протяжении 8 недель, при его наличии — 12 недель в целях повышения частоты излечения хронической инфекции.
Для удобства пациентов отдельные бемнифосбувир и рузасвир объединены в одну таблетку с фиксированной дозой.
ЧТО ЕЩЕ
В середине сентября 2024 года бемнифосбувир не справился с клиническим исследованием SUNRISE-3 (NCT05629962) фазы III среди амбулаторных высокорисковых пациентов с легко-умеренной коронавирусной инфекцией COVID-19: назначение препарата два раза в день на протяжении пяти дней поверх стандартного лечения не обеспечило статистически значимого снижения риска госпитализации или смерти по прошествии 29 дней [1].
БИЗНЕС
Период защиты интеллектуальной собственности «Атиа» заявлен вплоть до 2042 года — с потенциальной возможностью ее продления.
«Атиа», уже заплатившая «Мерк и Ко» аванс за лицензию на рузасвир, обязуется выдавать ей определенные суммы по мере прохождения этапов разработки, а также отчислять роялти от реализации готового препарата.
«Атиа» была основана в 2014 году Жаном-Пьером Соммадосси (Jean-Pierre Sommadossi) [1], который в свое время учредил «Айденикс фармасьютикалс» (Idenix Pharmaceuticals) и был соучредителем «Фармассет» (Pharmasset): первое предприятие в августе 2014 года купила «Мерк и Ко» за $3,85 млрд [2], вторую компанию в январе 2012 года приобрела «Гилеад сайенсиз» (Gilead Sciences) за $11,2 млрд [3].
В конце мая 2023 года «Атиа» отвергла предложение «Концентра байосайенсиз» (Concentra Biosciences), пожелавшей ее поглотить за $480 млн [4].