РЕЗЮМЕ
→ Немышечно-инвазивный рак мочевого пузыря (НМИРМП) встречается в три четверти случаев онкологического заболевания этого органа.
→ Первоочередное лечение НМИРМП предполагает трансуретральную резекцию (ТУР) всей видимой опухоли.
→ В целях профилактики рецидива назначают продолжительную иммунотерапию бациллой Кальметта — Герена (БЦЖ), которая весьма эффективна.
→ Однако в ряде случаев НМИРМП перестает реагировать на БЦЖ-вакцину, и тогда варианты дальнейшего консервативного лечения становятся резко ограниченными.
→ Фармотрасль усиленно работает над новыми способами лечения НМИРМП после провала БЦЖ-иммунотерапии.
СУТЬ ВОПРОСА
Во всём мире рак мочевого пузыря занимает десятое место среди наиболее часто диагностируемых видов онкологических заболеваний [1] [2]. Это онкологическое заболевание главным образом пожилых людей: средний возраст постановки диагноза составляет 69–71 лет [3].
В развитых странах рак мочевого пузыря преимущественно (в 90% случаев) характеризуется уротелиальной гистологией (ранее относился к переходно-клеточному раку) [4].
Уротелиальная карцинома мочевого пузыря в 70% случаев классифицируется как немышечно-инвазивный рак мочевого пузыря (НМИРМП), то есть как рак мочевого пузыря без прорастания в мышечный слой [5] [6].
После трансуретральной резекции (ТУР) всей видимой опухоли, как первоочередного хирургического лечения НМИРМП, 40–80% пациентов сталкиваются с рецидивом в течение последующих 6–12 месяцев, а 10–25% — прогрессируют до мышечно-инвазивного, регионарного или метастатического заболевания [7].
Таким образом, существует необходимость в способах лечения, дополняющих ТУР и преследующих цель сдерживания заболевания.
СЕГОДНЯ
Немышечно-инвазивный рак мочевого пузыря (НМИРМП), согласно гистологическому стадированию в зависимости от характера роста и глубины инвазии, может быть неинвазивной папиллярной карциномой (Ta), карциномой in situ (CIS; Tis) и с распространением на субэпителиальную соединительную ткань (T1) — соответственно в 70%, 10% и 20% случаев [1] [2].
После трансуретральной резекции (ТУР) всей видимой опухоли НМИРМП применяют, для предупреждения его рецидива или прогрессирования, интравезикальную (внутрипузырную) терапию, которая обеспечивает высокую локальную концентрацию терапевтического препарата внутри мочевого пузыря, потенциально уничтожая оставшиеся жизнеспособные опухолевые клетки.
Интравезикальное введение бациллы Кальметта — Герена (БЦЖ) — живой аттенуированной формы Mycobacterium bovis, возбудителя туберкулеза у крупного рогатого скота, предложенное еще в 1972 году [3] [4], является стандартной адъювантной процедурой, дополняющей ТУР при НМИРМП [5] [6] [7].
В качестве альтернативы БЦЖ-вакцине широко используется ряд химиотерапевтических препаратов, в частности, митомицин (mitomycin), эпирубицин (epirubicin) и гемцитабин (gemcitabine).
Высокую эффективность БЦЖ-иммунотерапии не удалось превзойти никакому из изученных внутрипузырных препаратов [8] [9] [10] [11] [12] [13].
Противоопухолевое действие БЦЖ-вакцины многогранно и включает следующие предполагаемые механизмы: индукция инфильтрата мононуклеарных клеток, состоящего преимущественно из T-клеток CD4+ и макрофагов; повышение экспрессии интерферона гамма (IFNγ); усиление экспрессии интерлейкинов 1, 2, 6, 8 и 12 (IL-1, IL-2, IL-6, IL-8, IL-12), фактора некроза опухоли (TNF) и TNF-связанного индуцирующего апоптоз лиганда (TRAIL); прямое подавление роста опухоли [31] [32] [33] [34] [35] [36].
Внутрипузырные инстилляции БЦЖ-вакцины показаны при высокорисковом немышечно-инвазивном раке мочевого пузыря, под определение которого подпадают CIS (Tis) и высокозлокачественные опухоли Ta или T1. Она также является вариантом для определенных пациентов с промежуточным риском заболевания [14] [15] [16] [17]. Согласно метаанализам, поддерживающее лечение при помощи БЦЖ на протяжении не менее чем одного года снижает риск рецидива или прогрессирования по сравнению с химиотерапией [18] [19] [20] [21].
БЦЖ-иммунотерапия способствует отсрочиванию прогрессирования опухоли до более распространенной стадии, снижению риска необходимости в последующей радикальной цистэктомиии (хирургическом удалении мочевого пузыря) и продлению общей выживаемости [19] [20] [21] [22] [23] [24] [25].
Однако в 40–50% случаев регистрируется провал БЦЖ-иммунотерапии, то есть, если упрощенно, обнаружение высокозлокачественной опухоли в ходе лечения или после него [18] [26] [27] [28] [29].
Дальнейшее лечение НМИРМП обычно предполагает радикальную цистэктомию, которая, сопровождающаяся заболеваемостью и смертностью, резко ухудшает качество жизни [30].
Вот почему необходимы варианты лечения немышечно-инвазивного рака мочевого пузыря, которые могли бы применяться после неудачи БЦЖ-иммунотерапии и которые приводили бы к устранению или отсрочиванию необходимости в радикальной цистэктомии.
НОВИНКИ
В начале января 2020 года «Китруда» (Keytruda, пембролизумаб), блокатор PD-1 авторства «Мерк и Ко» (Merck & Co.), получил разрешение Управления по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) для лечения высокорискового немышечно-инвазивного рака мочевого пузыря (НМИРМП) с карциномой in situ (CIS) с папиллярными новообразованиями (или без них), не реагирующего на назначение бациллы Кальметта — Герена (БЦЖ), — у пациентов, не подходящих для прохождения цистэктомии или отказавшихся от нее.

«Китруда» как спасительное лечение рака мочевого пузыря
Пембролизумаб позволит избежать радикальной цистэктомии.
В середине декабря 2022 года FDA одобрило «Адстиладрин» (Adstiladrin, надофараген фираденовек) — генно-терапевтическое лечение, разработанное швейцарской «Ферринг фармасьютикалс» (Ferring Pharmaceuticals) для применения при таком же показании НМИРМП.
Надофараген фираденовек (nadofaragene firadenovec, rAd–IFN/Syn3) построен на основе нереплицирующегося рекомбинантного аденовирусного вектора (rAd), кодирующего ген интерферона альфа-2b (IFNα-2b) человека. Последний, выступая естественным регулятором активности иммунной системы, активирует транскрипцию и трансляцию генов, ответственных за продукцию определенных ферментов, подавление клеточной пролиферации, а также иммуномодулирующую активность, включая фагоцитарную активность макрофагов и усиление специфической цитотоксичности лимфоцитов в отношении клеток-мишеней [1] [2] [3] [4] [5].

«Адстиладрин»: генная терапия рака мочевого пузыря
Надофараген фираденовек позволит избежать радикальной цистэктомии.
В конце апреля 2024 года на сцену вышел «Анктива» (Anktiva, ногапендекин альфа инбакицепт), предложенный «Имьюнитибайо» (ImmunityBio) для лечения такой же категории пациентов с НМИРМП. «Анктива», в отличие от монотерапии «Китрудой» или «Адстиладрином», назначается совместно с БЦЖ.
Ногапендекин альфа инбакицепт (nogapendekin alfa inbakicept, N-803, ALT-803) представляет собой антительный слитый (гибридный) белок, составленный из мутированного (N72D) человеческого интерлейкина 15 (IL-15), Sushi-домена внеклеточной области альфа-субъединицы рецептора IL-15 (IL-15Rα), Fc-области иммуноглобулина G1 (IgG1). Получившийся гетеродимерный комплекс является суперагонистом IL-15, который полностью имитирует противоопухолевую эндогенную биологию интерлейкина 15, добавляя к нему стабильность и продленный период полувыведения. Связывание ногапендекина альфа инбакицепта с рецепторами IL-15, экспрессирующими на T-клетках CD4+ and CD8+ и естественных киллерных (NK) клетках, приводит к активации и экспансии T-клеточного иммунитета, причем без стимуляции иммуносупрессивных регуляторных T-клеток (Treg), сдерживающих иммунный ответ [6] [7] [8].
ЗАВТРА
Фармотрасль продолжает разрабатывать новые способы лечения немышечно-инвазивного рака мочевого пузыря (НМИРМП) высокого риска, на который не подействовала иммунотерапия бациллой Кальметта — Герена (БЦЖ). Цель прозрачна: предложить консервативное лечение, которое максимально длительно откладывает необходимость в радикальной цистэктомии.
В своем подавляющем большинстве экспериментальные препараты применяются интравезикально (внутрипузырно).
ХИМИОТЕРАПИЯ
В продолжающемся клиническом испытании SunRISe-1 (NCT04640623) фазы IIb, организованном «Янссен» (Janssen) в составе «Джонсон энд Джонсон» (Johnson & Johnson), экспериментальная система TAR-200 (в виде 5-см кренделька) для таргетного и продолжительного (приблизительно 3 недели) локального высвобождения химиотерапевтического гемцитабина (gemcitabine) в мочевом пузыре обеспечила полный ответ у 83% (71–91) пациентов. Вероятность длительности ответа (DOR) на протяжении 1 года составила 75% (50–88) [1] [2].
Перспективной выглядит химиотерапевтическая тройка из кабазитаксела (cabazitaxel), гемцитабина (gemcitabine) и цисплатина (cisplatin), внутрипузырное последовательное введение которых, проверенное в клиническом исследовании фазы I, установило частоту безрецидивной выживаемости (RFS) на протяжении 12 и 24 месяцев на уровне 83% и 64%, притом что эти показатели оказались еще лучше, 100% и 83%, при получении максимальных доз [3]. К концу 2024 года ожидается завершение клинического испытания NCT02202772 фазы II [4].
Не следует также забывать об альтернативе БЦЖ-вакцине, которая давно находится в дефиците, в лице химиотерапевтических гемцитабина (gemcitabine) и доцетаксела (docetaxel), применяемых интравезикально и последовательно. Так, частоты 12- и 24-месячной RFS при назначении БЦЖ составили 71% (64–78) и 69% (62–76), а при химиотерапии — 85% (78–91) и 81% (72–87) [5] [6]. Продолжается клиническое исследование BRIDGE (NCT05538663) фазы III, которое призвано раскрыть нюансы выбора между БЦЖ-иммунотерапией и химиотерапией в ходе первоочередного лечения немышечно-инвазивного рака мочевого пузыря [7].
ТАРГЕТНАЯ ТЕРАПИЯ
«Янссен» (Janssen) в составе «Джонсон энд Джонсон» (Johnson & Johnson) подтвердила терапевтическую состоятельность эрдафитиниба (erdafitinib), тирозинкиназного ингибитора рецепторов 1–4 фактора роста фибробластов (FGFR1–4), альтерации которых, будучи онкодрайверными, обнаруживаются в 50–80% случаев НМИРМП [1] [2].
Во-первых, клиническое исследование THOR-2 (NCT04172675) фазы II, сравнившее пероральный эрдафитиниб с интравезикальной химиотерапией, показало его превосходство в том, что касается продления выживаемости без прогрессирования (PFS) [3].
Во-вторых, TAR-210, экспериментальное устройство для внутрипузырного локального высвобождения эрдафитиниба на протяжении 3 месяцев, справилось с клиническим исследованием NCT05316155 фазы I, продемонстрировав, согласно промежуточным результатам, способность сдерживать рецидив [4] [5].
Сейчас пероральный «Балверса» (Balversa, эрдафитиниб) разрешен для лечения местнораспространенного или метастатического рака мочевого пузыря с чувствительными к нему FGFR3-альтерациями, прогрессировавшего после или во время хотя бы одной линии системной терапии. В планах «Джонсон энд Джонсон» стоит расширение спектра показаний «Балверсы».
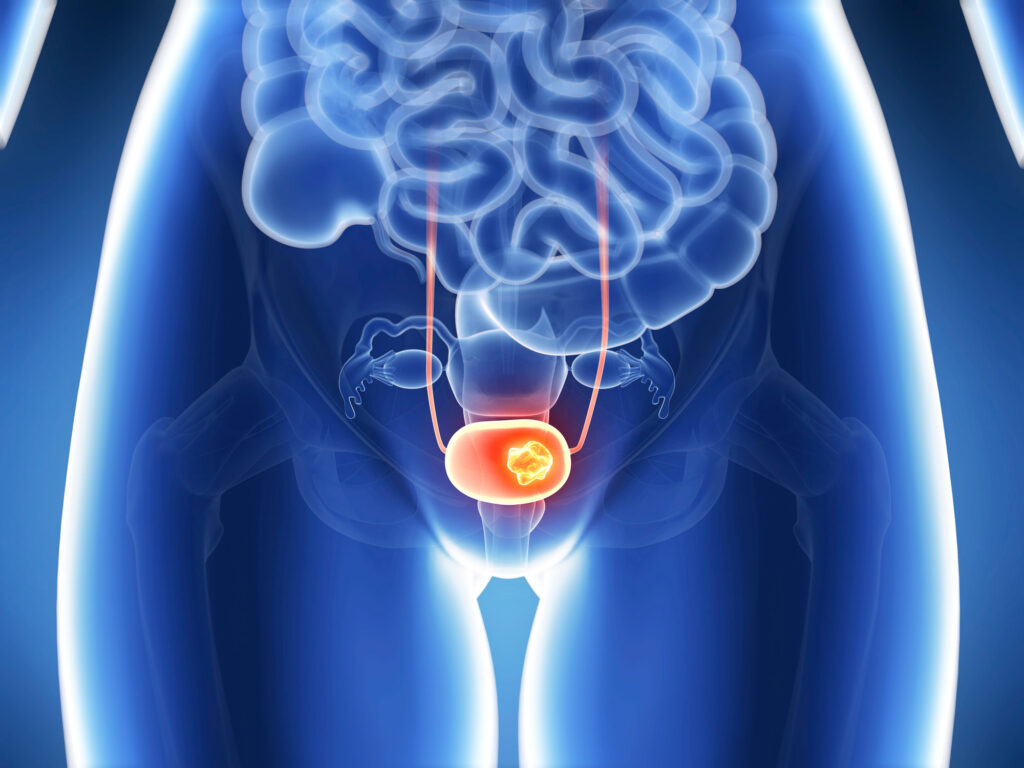
«Балверса»: таргетная терапия метастатического рака мочевого пузыря
Эрдафитиниб для лечения местнораспространенной или метастатической уротелиальной карциномы с FGFR3-альтерациями.
Промежуточные результаты клинического исследования EV-104 (NCT05014139) фазы I указали на потенциальную возможность интравезикального применения энфортумаба ведотина (enfortumab vedotin) в лечении НМИРМП после провала БЦЖ-иммунотерапии [6].
Этот конъюгат моноклонального антитела против нектина-4, известный как «Падцев» (Padcev) и несущий цитотоксический монометилауристатин E (MMAE), разработанный «Астеллас фарма» (Astellas Pharma) и «Сиджин» (Seagen), которую купила «Пфайзер» (Pfizer), сейчас назначается внутривенно в первоочередном лечении местнораспространенного или метастатического уротелиального рака.
«Падцев» + «Китруда»: более чем эффективное первоочередное лечение рака мочевого пузыря
Комбинация из энфортумаба ведотина и пембролизумаба продлит жизнь при неоперабельной уротелиальной карциноме.
Энфортумаб ведотин избирательно связывается с нектином-4 — иммуноглобулиноподобной молекулой клеточной адгезии и опухолеассоциированным антигеном, также известным как белок 4, связанный с рецептором полиовируса (PVRL4, PRR4), который сверхэкспрессирует на поверхности злокачественных клеток при различных солидных опухолях. После интернализации энфортумаба ведотина и протеолитического расщепления линкера происходит связывание MMAE с тубулином, что ингибирует полимеризацию последнего. Это приводит к остановке фазы G2/M клеточного цикла и индуцированию апоптоза опухолевых клеток, сверхэкспрессирующих нектин-4 [7] [8] [9] [10] [11].
Китайская «Римиджин» (RemeGen) сделала ставку на диситамаб ведотин (disitamab vedotin, RC48) — конъюгат моноклонального антитела, таргетированный на рецептор 2 эпидермального фактора роста 2 (HER2) и несущий цитотоксический монометилауристатин E (MMAE). Оценочная частота опухолевой HER2-экспрессии (совокупно IHC 2+ и 3+) при немышечно-инвазивном раке мочевого пузыря может доходить до 50% [12].
Сейчас этот препарат, одобренный под брендовым названием «Айдикси» (Aidixi), применяется внутривенно в лечении HER2-положительного местнораспространенного или метастатического рака желудка и уротелиального рака, ранее прошедших терапию. В августе 2021 года «Сиджин» приобрела у «Римиджин» мировые права (за исключением Азиатско-Тихоокеанского региона) на диситамаб ведотин [13].
Согласно ретроспективным клиническим данным, среди пациентов, получивших интравезикальные инстилляции диситамаба ведотина, частота 12-месячной RFS составила 100%, тогда как среди больных, которые прошли БЦЖ-иммунотерапию, этот показатель получился равным 58%. Впрочем, статистически значимого расхождения не зафиксировано (p=0,22) [14].
Запущены клинические исследования NCT06378242 фазы I/II, NCT05957757 фазы II и NCT05943379 фазы II, которые проверяют соответственно мононазначение диситамаба ведотина, его комбинацию с PD-1-блокатором «Тевимбра» (Tevimbra, тислелизумаб) авторства китайской «Бейджин» (BeiGene) и его сочетание с гемцитабином (gemcitabine). Первое испытание организовано среди пациентов, прежде не получавших БЦЖ-вакцину, второе и третье — уже прошедших ее курс.
Экспериментальная фотодинамическая терапия «Рутеррин» (Rutherrin), обкатываемая канадской «Тералейз текнолоджис» (Theralase Technologies) в ходе клинического исследования NCT03945162 фазы II, продемонстрировала, согласно промежуточным данным, вывод 63% (n=38/59) пациентов к статусу CR [15].
После интравезикального введения «Рутеррина», который сочетает производное рутения TLD-1433 с гликопротеином трансферрином, осуществляется его активация лазерным излучением. «Рутеррин», преимущественно поглощенный раковыми клетками (они характеризуются повышенной экспрессией рецепторов трансферрина), начинает вырабатывать синглетный (атомарный) кислород и кислородные радикалы — активные формы кислорода, вызывающие в этих клетках окислительный стресс с последующей их гибелью [16] [17] [18] [19] [20] [21].
ИММУНОТЕРАПИЯ
Продолжается клиническое исследование HOPE-04 (ChiCTR2200059970) фазы II, проверяющее гипотезу оправданности короткого курса облучения в сочетании с внутривенным торипалимабом (toripalimab), блокатором PD-1 китайской «Шанхай Цзюньши байосайенсиз» (Shanghai Junshi Biosciences), одобренным под брендом «Локторзи» (Loqtorzi) для лечения рака носоглотки [1].
Китайская «Цзянсу Симсиа фармасьютикал» (Jiangsu Simcere Pharmaceutical) придумала добавлять к БЦЖ-вакцине (или использовать монотерапевтически) экспериментальный противоопухолевый и иммуностимулирующий слитый белок SIM0237, составленный из моноклонального антитела против PD-L1 и комплекса со сниженной потентностью, включающего интерлейкин 15 (IL-15) и Sushi-домен внеклеточной области альфа-субъединицы рецептора IL-15 (IL-15Rα) [2] [3]. Клиническое исследование NCT06186414 фазы II продолжается.
БАКТЕРИОТЕРАПИЯ
Экспериментальный TARA-002, изучаемый «Протара терапьютикс» (Protara Therapeutics), продемонстрировал обнадеживающие 3-месячные результаты клинических исследований ADVANCED-1 (NCT05085977) фазы Ia/Ib и ADVANCED-2 (NCT05951179) фазы II: совокупный выход к CR-статусу составил 38% (n=6/16), варьируя в широких пределах в зависимости от особенностей заболевания и его предшествовавшего лечения [1].
TARA-002 — иммунопотенцирующий препарат широкого спектра действия, по своему механизму действия схожий с БЦЖ-вакциной, активирующий врожденные и адаптивные иммунные клетки, а также непосредственно уничтожающий опухолевые клетки. TARA-002 разработан на основе того же клеточного банка из генетически разнообразных пиогенных стрептококков (Streptococcus pyogenes) группы A, что и «Пицибанил» (Picibanil, OK-432), продвигаемый японской «Шугай фармасьютикал» (Chugai Pharmaceutical) и успешно применяющийся в Японии и на Тайване с 1975 года для лечения лимфангиомы (лимфатической мальформации) [2] [3] [4] [5].
В начале февраля 2024 года началось клиническое исследование PARADIGM-1 (NCT06181266) фазы I/Ib, тестирующее экспериментальный ZH9, который британская «Прокариум» (Prokarium) создала на основе генетически модифицированного штамма ZH9 аттенуированной бактерии Salmonella enterica подвида enterica серовара Typhi, вызывающей брюшной тиф [6] [7].
По сути «Прокариум», ратующая за идею альтернативы БЦЖ-вакцине, лицензировала наработки Университетской больницы Лозанны (CHUV) [8], специалисты которого неоднократно демонстрировали терапевтическую оправданность данного подхода на примере интравезикальной рецептуры пероральной тифозной вакцины «Вивотиф» (Vivotif, Ty21a) [9] [10] [11] [12] [13].
ВИРУСНАЯ ТЕРАПИЯ
«Си-джи онколоджи» (CG Oncology) предложила атаковать НМИРМП онколитическим вирусом кретостимоген гренаденорепвек (cretostimogene grenadenorepvec, CG0070), который избирательно реплицируется в опухолевых клетках с дефектом сигнального пути белка ретинобластомы (RB). При раке мочевого пузыря его опухолевые клетки почти всегда несут RB-мутации. Кретостимоген гренаденорепвек работает путем прямого лизиса опухоли и через механизмы иммуногенной гибели раковых клеток [1] [2] [3] [4] [5].
Согласно промежуточным данным клинических испытаний, кретостимоген гренаденорепвек предоставил лучшие исходы, если говорить о частоте полного ответа (CR) — в сравнении с уже одобренными препаратами против НМИРМП, такими как «Китруда» (Keytruda, пембролизумаб), «Адстиладрин» (Adstiladrin, надофараген фираденовек) и «Анктива» (Anktiva, ногапендекин альфа инбакицепт).
«Трувезик» / «Арамира»: иммуноонкологическое лечение рака мочевого пузыря
Кретостимоген гренаденорепвек: онколитический вирус против уротелиальной карциномы, не реагирующей на иммунотерапевтическую вакцину БЦЖ.
ГЕННАЯ ТЕРАПИЯ
Экспериментальная невирусная иммуноонкологическая генная терапия деталимоген вораплазмид (detalimogene voraplasmid, EG-70) выдала CR на уровне 73% (n=16/22) в продолжающемся клиническом исследовании LEGEND (NCT04752722) фазы I/II [1] [2].
Деталимоген вораплазмид, разработкой которого занимается американо-канадская «Энджин» (enGene), доставляет в эпителий слизистых тканей мочевого пузыря ДНК-плазмиду для локальной экспрессии рекомбинантного одноцепочечного интерлейкина 12 (IL-12) [двух его субъединиц, p40 и p35 ] и двух двухцепочечных РНК-активаторов (VA1 и eRNA11a) сигнального пути индуцируемого ретиноевой кислотой гена 1 (RIG-I), являющегося внутриклеточным регулятором врожденного иммунитета.
Механизм действия деталимогена вораплазмида обращается к индуцированию выработки интерферона гамма (IFNγ), который является сильным противоопухолевым и антиангиогеннным цитокином, и стимулированию мощного воспалительного ответа, который приводит к прямому уничтожению опухолевых клеток, опосредованной цитокинами активации клеток врожденного иммунитета, рекрутингу и перекрестному примированию T-клеток.
РНК-ТЕРАПИЯ
В конце апреля 2024 года китайская «Рактиджен терапьютикс» (Ractigen Therapeutics) получила разрешение Управления по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) на проведение первого клинического испытания экспериментального RAG-01, который построен на базе коротких активирующих РНК (каРНК) и который восстанавливает экспрессию опухолевого супрессора p21, обычно заглушенного в раковых клетках мочевого пузыря [1].
ПРОВАЛЫ
Определенных успехов в лечении высокорискового немышечно-инвазивного рака мочевого пузыря (НМИРМП), который перестал реагировать на иммунотерапию бациллой Кальметта — Герена (БЦЖ), добилась «Сесен байо» (Sesen Bio), ранее называвшаяся «Илевен байотерапьютикс» (Eleven Biotherapeutics) и в марте 2023 года слившаяся с «Карисма терапьютикс» (Carisma Therapeutics) [1].
Экспериментальный «Вицинеум» (Vicineum, опортузумаб монатокс) по прошествии 3 месяцев клинического исследования VISTA (NCT02449239) фазы III вывел 40% (30–51) пациентов с к полному ответу (CR), хотя, когда миновали 12 месяцев, ремиссия сохранилась лишь у 17% (10–26) испытуемых [2].
Опортузумаб монатокс (oportuzumab monatox, VB4-845) — иммунотоксин, состоящий из гуманизированного одноцепочечного вариабельного фрагмента моноклонального антитела против молекулы клеточной адгезии эпителия (EpCAM), конъюгированного с усеченной формой экзотоксина A синегнойной палочки (Pseudomonas aeruginosa). Поскольку при раке мочевого пузыря наблюдается сверхэкспрессия мембранного белка EpCAM, опортузумаб монатокс осуществляет таргетную доставку в опухолевые клектки цитотоксической нагрузки, которая их уничтожает, блокируя белковый синтез [3] [4] [5] [6].
В июле 2022 года «Сесен» свернула программу разработки «Вицинеума» для лечения НМИРМП [7].
Не оправдала себя затея объединения БЦЖ-вакцины с противораковой вакциной «Панвак» (Panvac): исходы клинического исследования NCT02015104 фазы II статистически значимо не отличались от применения только БЦЖ-иммунотерапии [8].
Онковакцина «Панвак» (CEA-MUC-1-TRICOM, CV301, inalimarev–falimarev), разработанная «Терион байолоджикс» (Therion Biologics), состоит из двух векторов: модифицированного осповакцинного вируса и рекомбинантного вируса оспы кур, — они применяются в режиме прайм-буст. Оба вектора кодируют трансгены двух опухолеассоциированных антигенов, муцина 1 (MUC1) и карциноэмбрионального антигена (CEA), и трех костимулирующих T-клетки молекул, B7.1 (CD80), ICAM-1 (CD54) и LFA-3 (CD58). За счет того, что антигенпрезентирующим клеткам (APC) демонстрируются MUC1 и CEA, которые сверхэкспрессированы при карциномах, активируется ответ цитотоксических T-лимфоцитов (CTL). В качестве опционального адъюванта используется гранулоцитарно-макрофагальный колониестимулирующий фактор (GM-CSF) [9] [10] [11] [12] [13] [14].
Не повезло израильской «Анкиано терапьютикс» (Anchiano Therapeutics), в марте 2021 года объединившейся с местной «Кемоумаб» (Chemomab) и теперь называющейся «Кемоумаб терапьютикс» (Chemomab therapeutics): в ноябре 2019 года пришлось остановить программу весьма интересной генной терапии, но не обеспечившей должной частоты ремиссии. В клиническом исследовании CODEX (NCT03719300) фазы II, согласно промежуточному анализу, к CR-статусу вышли 19% (n=3/16) пациентов [15] [16].
Инодифтаген викстеплазмид (inodiftagene vixteplasmid, BC-819) — рекомбинантная ДНК-плазмида, несущая ген цепи дифтерийного токсина A (dT-A) и протомор H19. За счет того, что транскрипционные факторы H19 в избытке представлены в опухолевых клетках, осуществляется активация экспрессии dT-A, который подавляет синтез белков и вызывает гибель опухолевых клеток. Инодифтаген викстеплазмид не содержит гена цепи дифтерийного токсина B (dT-B), что предотвращает передачу цепи dT-A между клетками. Онкофетальный ген H19 высоко экспрессируется эмбриональными и некоторыми злокачественными тканями, но слабо представлен в нормальных взрослых тканях [17] [18] [19] [20] [21].