«Илай Лилли» (Eli Lilly) надеется к 2026 году вывести на рынок орфорглипрон (orforglipron) — пероральный лекарственный препарат, предназначенный для долгосрочного снижения веса.
ОСНОВНЫЕ ФАКТЫ
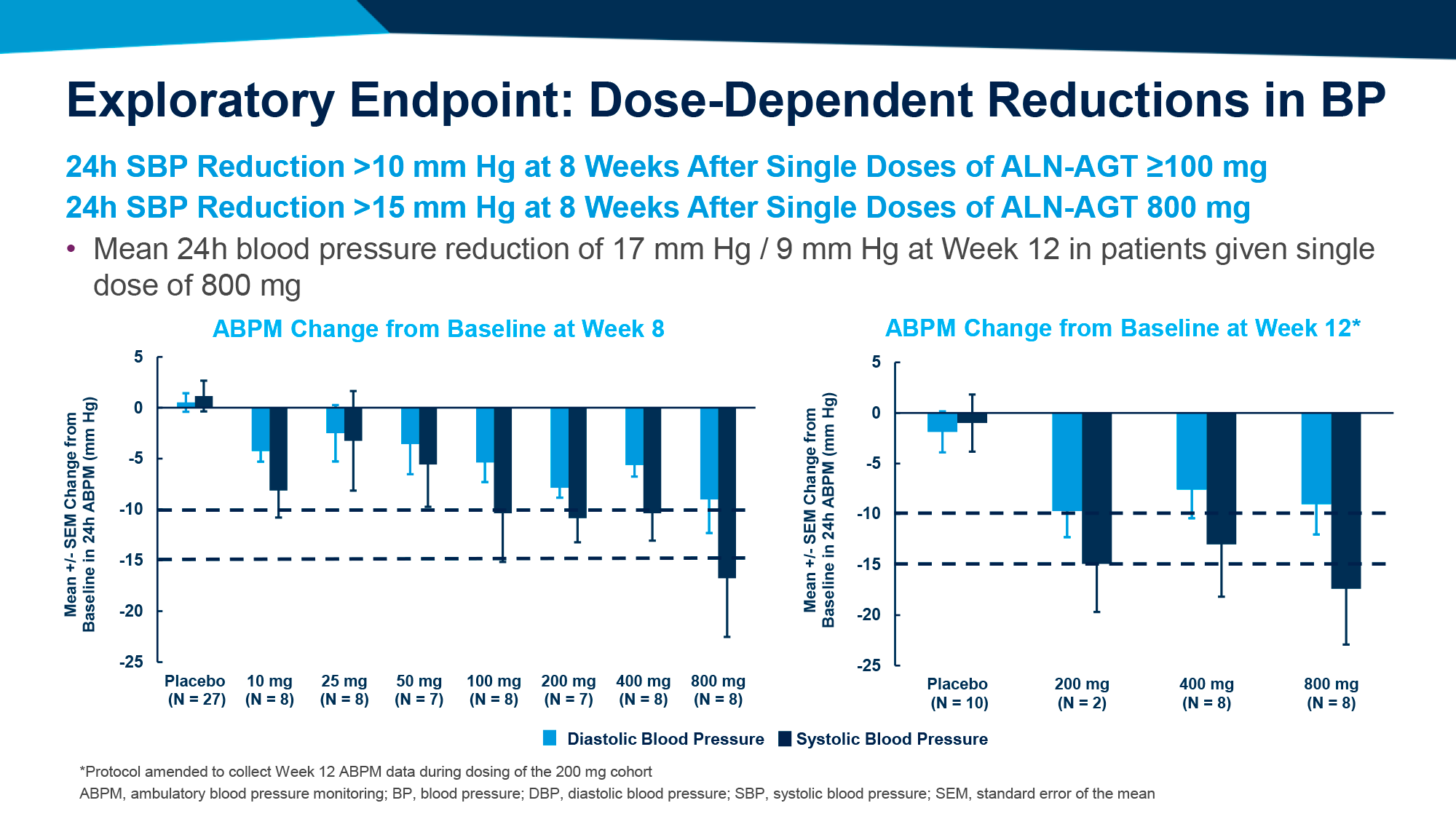
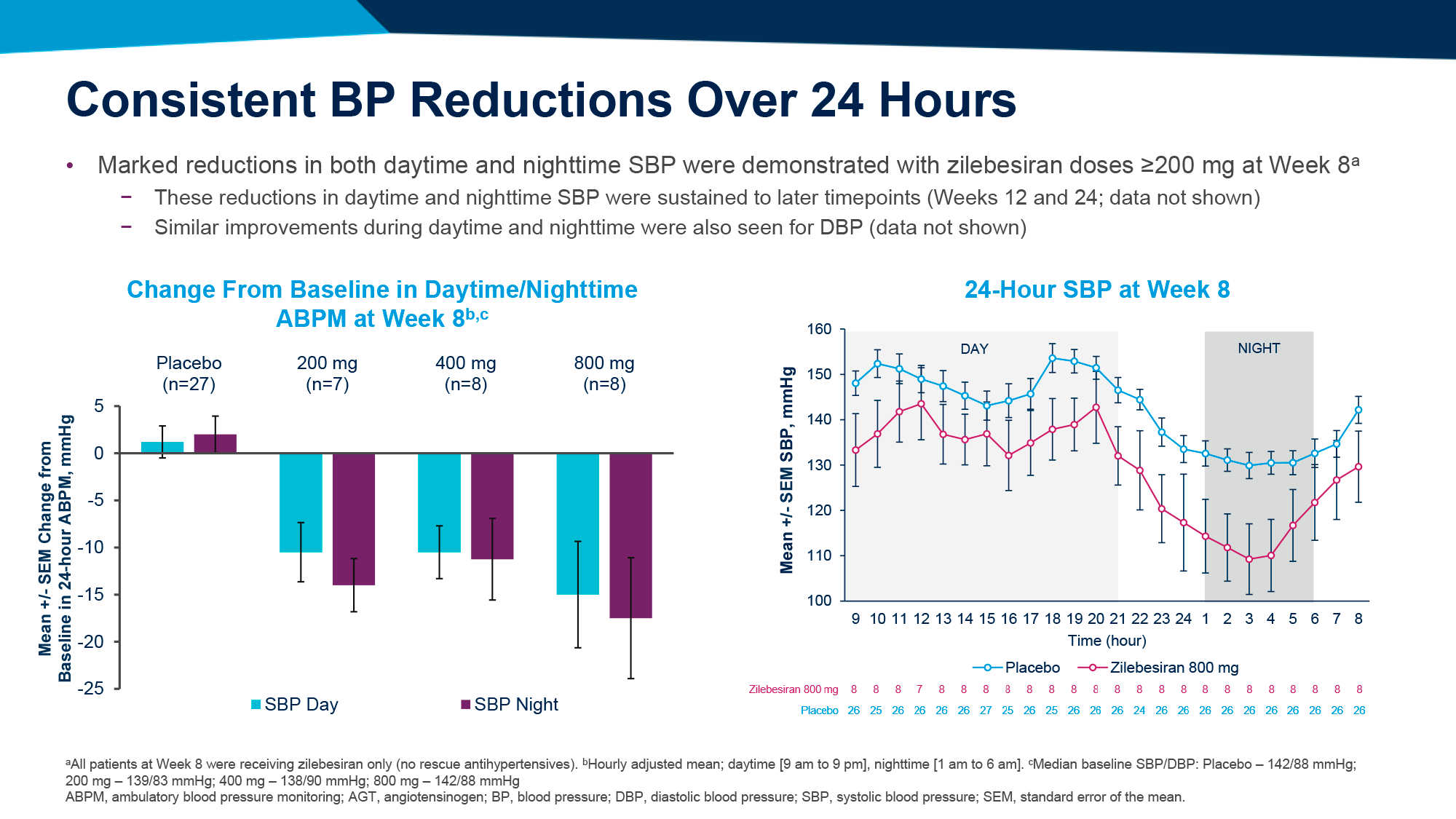
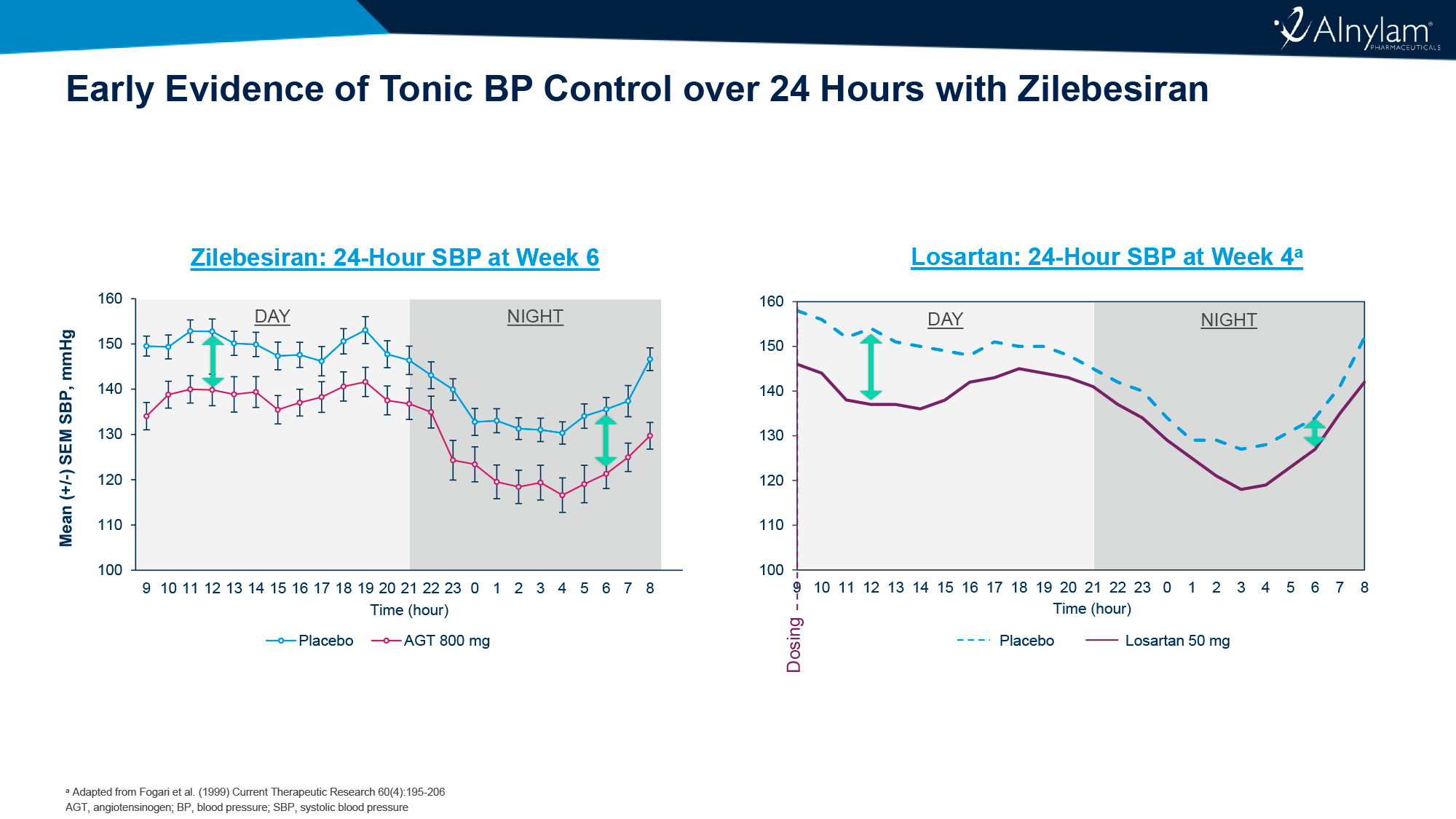
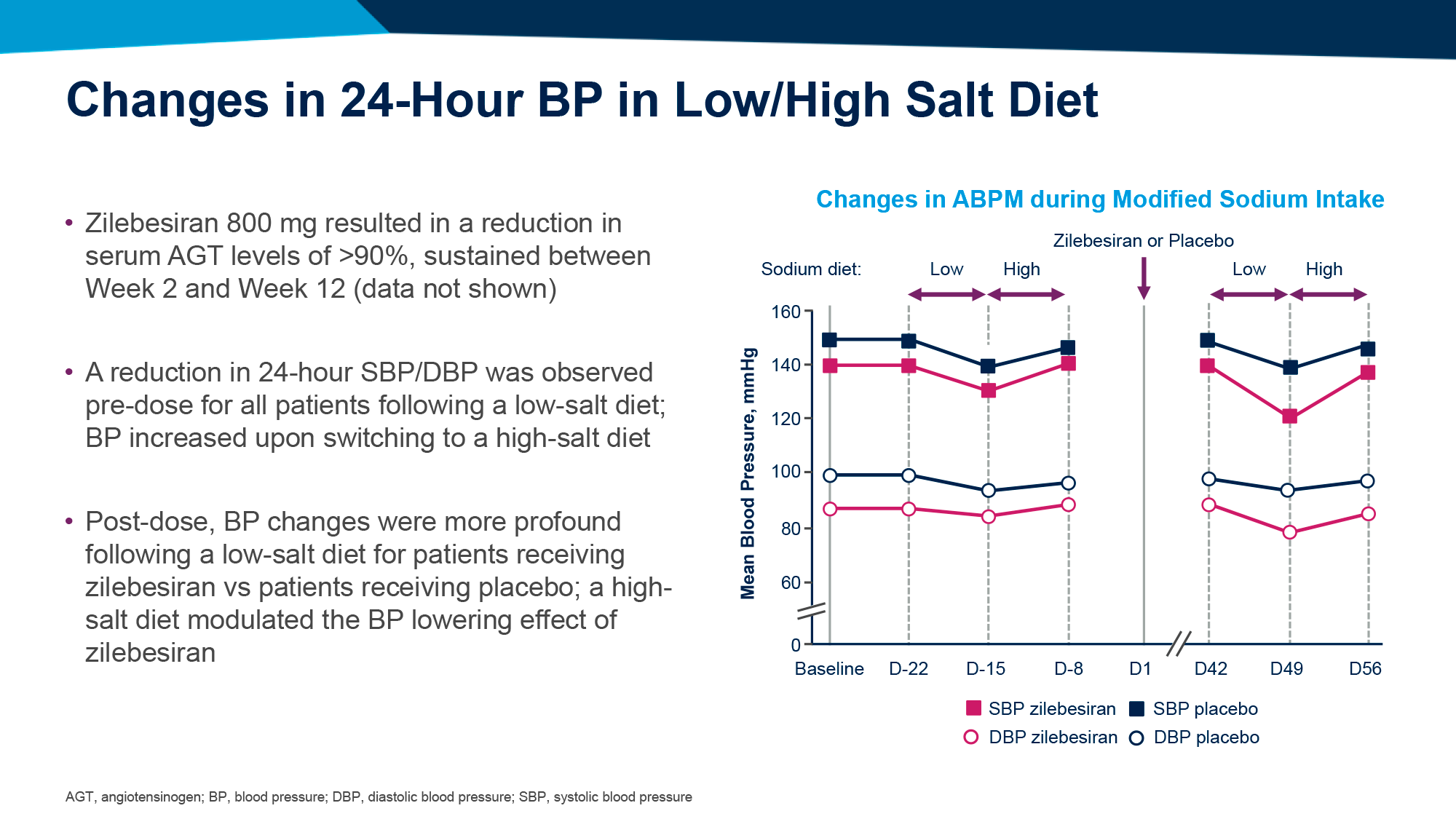
Среднестадийная клиническая проверка установила, что ежедневное назначение орфорглипрона привело к похудению на 8,6–12,6% (9,0–13,3 кг) за 26 недель лечения и на 9,4–14,7% (9,8–15,4 кг) — за 36 недель.
Орфорглипрон, прилично снизивший уровень глюкозы в крови, доказал собственную пригодность в терапии сахарного диабета 2-го типа (СД2).
Орфорглипрон, если справится с масштабными клиническими испытаниями фазы III, поступит в продажу в 2026 году.
Орфорглипрон, будучи непептидным низкомолекулярным соединением, относится к классу агонистов рецептора глюкагоноподобного пептида-1 (GLP1RA) — мегапопулярных препаратов, повсеместно применяемых для борьбы с лишним весом.
Согласно отраслевым прогнозам, орфорглипрон мгновенно станет бестселлером, зарабатывающим многие миллиарды долларов. Грядущий успех обеспечен равно как приемлемой терапевтической эффективностью, так и удобством таблеточной реализации, не зависящей от приема пищи и не требующей холодовой цепи для хранения и логистики.
Для «Илай Лилли», уже располагающей современными инъекционными GLP1RA-препаратами в лице «Мунджаро» (Mounjaro, тирзепатид) и «Зепбаунда» (Zepbound, тирзепатид), чрезвычайно важно усилить собственное присутствие на рынке лекарств для похудения. Ведь конкуренция нагнетается со страшной силой: десятки фармкомпаний изобретают всё новые лекарства для желающих сбросить вес.
Не исключено, орфорглипрон займет должное место в хронической поддерживающей терапии, когда лишняя масса тела уже устранена мощным тирзепатидом (tirzepatide), но необходимо придерживаться долгосрочного фармакологического контроля над аппетитом, чтобы вес не вернулся.
Тем временем «Ново Нордиск» (Novo Nordisk) готовится раскрыть новые результаты применения перорального семаглутида (semaglutide) для похудения. Его регуляторное одобрение ожидается в 2025 году.
К чему изобретать своё, если можно взять готовое? А ты, Eli Lilly, страшись!
ПРЯМАЯ РЕЧЬ
«Ожирение, будучи глобальной эпидемией, давно нуждается в различных эффективных препаратах, которые различаются по своему способу применения. Орфорглипрон демонстрирует превосходные результаты, притом что он назначается без каких-либо ограничений по приему пищи или жидкости».
Шон Уортон (Sean Wharton), директор медицинской клиники Уортона (Онтарио, Канада), специализирующей на лечении ожирения.
«Люди, страдающие хроническими заболеваниями, такими как ожирение и сахарный диабет 2-го типа, заслуживают того, чтобы у них были варианты эффективного лечения, в том числе в виде удобных пероральных препаратов».
Джефф Эммик (Jeff Emmick), старший вице-президент по разработке продукции «Илай Лилли» (Eli Lilly).
КАК ЭТО РАБОТАЕТ
GLP1RA-препараты — нынешний фармакологический стандарт лечения ожирения, а также избыточной массы тела при наличии сопутствующих лишнему весу заболеваний.
GLP1RA-препараты имитируют глюкагоноподобный пептид-1 (GLP-1) — инкретиновый гормон, который способствует снижению веса, уменьшая аппетит и задерживая опорожнение желудка, что по итогам приводит к улучшению энергетического баланса [1].
Орфорглипрон (orforglipron, LY3502970, OWL833) представляет собой пероральный непептидный низкомолекулярный GLP1RA. Все одобренные на сегодня GLP1RA-препараты против ожирения реализованы инъекционными пептидными лекарственными средствами.
«Ребелсас» (Rybelsus, семаглутид), предложенный «Ново Нордиск», является пока единственным на рынке пероральным GLP1RA-препаратом. Он одобрен для терапии СД2, а его повышенные дозы изучаются в задаче снижения веса. Пероральный семаглутид необходимо принимать хотя бы за 30 минут до еды: ввиду низкой биодоступности этого пептидного лекарства, рецептура которого подкреплена усилителем проницаемости салкапрозатом натрия (SNAC) [2], существует риск снижения терапевтического эффекта.
Novo Nordisk выпустила семаглутид в виде таблеток.
Механизм активации рецептора GLP-1, осуществляемый орфорглипроном, таков, что рождающаяся в ответ на его применение сигнализация циклического аденозинмонофосфата (цАМФ) весьма схожа с сигналом, производимым нативным GLP-1. Благодаря тому, что орфорглипрон не вызывает сильной активации бета-аррестинового сигнального пути, который регулирует интернализацию рецептора, результирующая терапевтическая эффективность получается приемлемой [3].
Подобный смещенный агонизм GLP-1 характерен для тирзепатида, который весьма успешно и превосходящим семаглутид образом снижает вес [4].
КЛИНИЧЕСКИЕ ПОДРОБНОСТИ
ОЖИРЕНИЕ
Клиническое исследование NCT05051579 фазы II (рандомизированное, двойное слепое, плацебо-контролируемое, многоцентровое, международное) проверило ежедневное пероральное назначение орфорглипрона (в разных дозах, с постепенным повышением дозы до целевой) среди взрослых пациентов (n=272), страдающих либо ожирением (индекс массы тела [ИМТ] ≥ 30 кг/м2), либо лишним весом (ИМТ ≥ 27 и <30 кг/м2) с сопутствующим ему расстройством (гипертония, дислипидемия или сердечно-сосудистое заболевание).
Испытуемым было рекомендовано придерживаться диеты и повышенной физической нагрузки.
По прошествии 26 недель лечения участники, получавшие орфорглипрон в дозе 12, 24, 36 или 45 мг, продемонстрировали снижение массы тела на 8,6% (95% ДИ [здесь и далее]: 6,9–10,2), 11,2% (9,6–12,8), 12,3% (10,7–13,8) и 12,6% (11,1–14,1) — против уменьшения веса на 2,0% (0,4–3,6) в группе плацебо. Это эквивалентно потери 9,0 (7,2–10,7), 12,3 (10,6–14,0), 12,9 (11,3–14,5) и 13,3 (11,7–14,9) кг — против 2,1 (0,4–3,8) кг [1] [2].
По истечении 36 недель терапии процесс похудения продолжился: вес снизился на 9,4% (7,4–11,5), 12,5% (10,5–14,5), 13,5% (11,6–15,3) и 14,7% (12,8–16,5) — против потери 2,3% (0,4–4,3). В абсолютном выражении уменьшение веса составило 9,8 (7,6–11,9), 13,6 (11,6–15,7), 14,2 (12,3–16,2) и 15,4 (13,5–17,4) кг — против 2,4 (0,4–4,5) кг.
Как минимум 5% лишнего веса сбросили 72%, 90%, 92% и 90% пациентов — против 24%. Не менее чем на 10% похудели 46%, 62%, 75% и 69% испытуемых — против 9%. Избавились хотя бы от 15% веса 22%, 33%, 43% и 48% — против 1%.
Соответственно избавлению от жировых отложений улучшились кардиометаболические показатели, включая уровни гликированного гемоглобина (HbA1C), холестерина, триглицеридов, АЛТ, АСТ, артериального давления.
Назначение орфорглипрона привело к снижению уровней биомаркеров воспаления и сердечно-сосудистого риска, таких как интерлейкин 6 (IL-6), высокочувствительный C-реактивный белок (hsCRP), аполипопротеин B (ApoB), аполипопротеин C3 (ApoCIII), лептин.
Тошнота, запор, рвота, диарея и отрыжка — наиболее частые нежелательные явления (НЯ) в ответ на назначение орфорглипрона. В большинстве случаев эти НЯ носили легко-умеренную степень выраженности, возникали в период постепенного повышения дозы, были преходящими и разрешались самостоятельно.
Еженедельные инъекции тирзепатида — минус 24 килограмма за полтора года.
ДИАБЕТ
Клиническое исследование NCT05048719 фазы II (рандомизированное, двойное слепое, плацебо-контролируемое, многоцентровое, международное) изучило ежедневное пероральное применение орфорглипрона (в разных дозах, с постепенным повышением дозы до целевой) среди взрослых пациентов (n=383) с СД2.
Среди основных требований к испытуемым: следование диете и повышенной физической нагрузке, опциональный прием метформина, уровень HbA1C 7,0–10,5%.
На протяжении 26 недель участникам назначали перорально орфорглипрон (3, 12, 24, 36 или 45 мг один раз в день), инъекционно дулаглутид (dulaglutide) [1,5 мг еженедельно] или плацебо.
В группах, получавших орфорглипрон, зарегистрировано снижение уровня HbA1C на 1,19%, 1,91%, 1,79%, 2,03% и 2,10% — против его снижения на 1,10% и 0,43% в группах дулаглутида и плацебо [1] [2] [3].
Уровень глюкозы натощак упал на 32,6; 53,7; 52,2; 53,9 и 55,9 мг/дл — против его падения на 33,2 и 11,1 мг/дл.
Масса тела уменьшилась на 3,8% (3,7 кг), 6,5% (6,5 кг), 9,9% (9,7 кг), 9,6% (9,5 кг) и 9,6% (10,1 кг) — против ее уменьшения на 3,9% (3,9 кг) и 2,2% (2,2 кг).
Среди прочих благотворных при СД2 эффектов орфорглипрона: улучшение бета-клеточной функции и чувствительности к инсулину.
Спектр и частота НЯ носили привычный для GLP1RA-препаратов характер.
ЧТО ДАЛЬШЕ
Орфорглипрон продолжает проходить глобальную регистрационную клиническую программу фазы III, включающую испытания ATTAIN против ожирения и лишнего веса и ACHIEVE — СД2. Исследования, охватывающие свыше 11 тыс. человек, должны завершиться ко второй половине 2025 года.
ЭКСПЕРТНЫЕ КОММЕНТАРИИ
Максимальная разница с плацебо, которую выдал орфорглипрон в отношении снижения веса, составила 12,4% за 36 недель терапии. Да, эффект уступает результатам применения того же тирзепатида, но весьма приемлем, если сравнивать его с пероральным семаглутидом, ежедневно назначаемым в дозе 50 мг и обеспечившим максимальную разницу с плацебо на уровне 12,7% (или 15,6%, если пациенты безоговорочно следовали протоколу лечения), однако за долгих 68 недель. Опять же, не исключено, процесс похудения на орфорглипроне к своему плато еще не вышел, и дальнейшее неспешное уменьшение массы тела продолжится.
Одна таблетка в день — минус 18 кг за полтора года.
Терапевтический эффект орфорглипрона не зависит от приема пищи, и это уже дополнительный плюс в сравнении с пероральным семаглутидом [1].
Желудочно-кишечные побочные эффекты, расхожие при назначении GLP1RA-препаратов, всё также присутствуют. Возможно, их частоту удастся снизить путем иной, более плавной схемы повышения дозы (начиная с меньшей дозы и следуя медленной ее эскалации) — это станет понятно по завершении клинической проверки фазы III. Впрочем, рассчитывать на какие-то серьезные улучшения переносимости вряд ли стоит, потому что на деле всё упирается в период полувыведения: чем он короче, тем более выражены пики и впадины в фармакокинетике.
Поскольку орфорглипрон является малой молекулой, его выпуск не сопряжен со значительными трудностями и ограничениями мощностей, как в случае нынешних инъекционных GLP1RA-препаратов, которые всё еще не избавились от проблем дефицита ввиду высокого спроса. Нельзя сказать, что себестоимость производства орфорглипрона копеечная, ведь эта структурно сложная молекула требует, есть мнение, не менее трех десятков этапов химического синтеза. Но при любом раскладе затраты не столь велики, если сравнивать с инъекционными лекарствами, требующими стерильных условий производства.
Сможет ли пероральный орфорглипрон составить серьезную конкуренцию инъекционным GLP1RA-препаратам? «Илай Лилли» уходит от прямого и однозначного ответа, ссылаясь на ряд предполагаемых факторов и особенностей рынка, прогнозируемо выгодных, по мнению американской фармкомпании, для бизнеса орфорглипрона [2] [3] [4]. Впрочем, приводимые доводы кажутся весьма натужными и не слишком убедительными для инвесторов.
Так, по собственным данным «Илай Лилли», приблизительно 20% пациентов с ожирением не хотят обращаться к инъекционным препаратам. Здесь, возможно, определенную роль играет страх перед подобным способом введения лекарства. Хотя, учитывая насколько удобной, безопасной и не вызывающей каких-либо неприятных ощущений является современная инъекционная инсулинотерапия, данное утверждение не выдерживает критики.
Фармацевтическое предприятие из Индианаполиса уверено, что на крупных азиатских рынках сбыта, включая Японию и Китай, пациенты отдают предпочтение пероральным способам лечения [5] [6] [7]. Но это зависит от методологии оценки.
Верным является утверждение, что в большинстве стран лечение ожирения не покрывается медицинской страховкой, то есть не бесплатно для конечного потребителя. Это обусловлено, скорее всего, предшествовавшим опытом, когда «старые» препараты для борьбы с лишним весом обеспечивали совсем незначительное его снижение, а их прием сопровождался неприемлемыми НЯ. Современные препараты вывели лечение ожирения на качественно новый уровень, поэтому в перспективе они всё же будут охвачены страховым покрытием, если учитывать ту массу преимуществ для здоровья, которое предоставляет эффективное избавление от жировых отложений. Однако, ввиду того, что процесс принятия данного решения затяжной, имеет смысл предложить пероральный GLP1RA-препарат с более приемлемой для пациентов ценовой доступностью в сравнении с инъекционными лекарствами. Всё, впрочем, упирается в конечную стоимость орфорглипрона.
Наконец, полностью соответствует истине тот факт, что, принимая во внимание огромное число людей в мире, страдающих от лишнего веса и ожирения (соответственно 2,5 млрд и 890 млн человек [8]), инъекционных GLP1RA-препаратов попросту не хватает на всех. Заводы загружены полностью, спрос только растет, ввод в строй новых производственных мощностей занимает продолжительное время. Так что для орфорглипрона обязательно найдется место.
Мощные и безопасные лекарства для похудения — всё только начинается.
ИЗ ИСТОРИИ
В сентябре 2018 года «Илай Лилли» приобрела у японской «Шугай фармасьютикал» (Chugai Pharmaceutical) мировые права на разработку и коммерциализацию орфорглипрона (OWL833). Авансом было выложено $50 млн и обещаны последующие отчисления от продаж готового препарата в размере от 4–6% до 10–13% в зависимости от объемов реализации. По итогам «Шугай» может получить до $140 млн по мере прохождения регуляторных этапов и до $250 млн по ходу продаж [1] [2].
Как видим, суммы решительно незначительные, учитывая будущий статус бестселлера. Но этому есть объяснение.
На момент покупки орфорглипрона он находился на доклинической стадии, а само направление GLP1RA-препаратов было развито очень слабо, равно как и не было абсолютно никакого потребительского ажиотажа вокруг фармакологического лечения ожирения: потому что предлагаемые препараты работали из рук вон плохо.
К слову, у «Рош» (Roche), которая два десятка лет тесно дружит с «Шугай», эксплуатируя ее глубочайшую научно-исследовательскую экспертизу, было «право преимущественного приобретения» орфорглипрона, но швейцарский фармацевтический гигант им не воспользовался, бросив все силы на развитие онкологического бизнеса.
Решение «Илай Лилли» поставить на орфорглипрон и профинансировать его доведение до ума наглядно демонстрирует, что крупнейшие игроки на рынке лечения СД2 располагают большим преимуществом в понимании особенностей метаболизма и потери веса, и потому способны принимать верные решения, пусть даже долгосрочные в своей конечной реализации.
«Рош» (Roche) всеми силами старается запрыгнуть в стремительно мчащийся экспресс лекарственных препаратов для похудения.
ОСНОВНЫЕ ФАКТЫ
«Рош» поставила перед собой задачу прямой конкуренции с «Ново Нордиск» (Novo Nordisk) и «Илай Лилли» (Eli Lilly), ведущими игроками на рынке современных лекарственных средств, предназначенных для долгосрочного снижения веса.
Первые препараты против ожирения выйдут из стен швейцарского фармацевтического гиганта ориентировочно в 2027–2028 гг.
Их разработка осуществляется благодаря приобретенной в конце января 2024 года «Кармот терапьютикс» (Carmot Therapeutics), за которую было выложено авансовых $2,7 млрд наличными с последующими потенциальными выплатами до $400 млн по мере развития лекарственных программ [1] [2].
Согласно промежуточным данным, лекарственные проекты «Кармот» характеризуются эффективностью и безопасностью, которые превосходят большинство нынешних препаратов, одобренных и экспериментальных.
Изучаемые молекулы относятся к классу агонистов рецептора глюкагоноподобного пептида-1 (GLP1RA) и агонистов рецептора глюкозозависимого инсулинотропного полипептида (GIPRA), вокруг которых построены почти все нынешние усилия фармацевтической отрасли, занимающейся вопросами ожирения.
Благодаря тому, что GLP1RA/GIPRA-препараты «Кармот» являются либо гибридными, сочетающими пептидные и низкомолекулярные фрагменты, либо чисто низкомолекулярными, они проще в исследовании и производстве, нежели полностью пептидные лекарства конкурентов. Однако они также менее предсказуемы, чем только пептидные, в том, что касается неожиданных побочных эффектов.
Между тем поздний вход «Рош» в бизнес лечения ожирения приведет к тому, что, согласно отраслевым прогнозам, она сможет заработать на этом лишь $4 млрд к 2032 году. И это крохи с учетом предполагаемого размера этого рынка в $150 млрд к 2033 году — против жалких $0,5 млрд в 2020-м.
Успехи современных препаратов против ожирения, способствующих снижению веса на 15–25% за год-полтора, отражаются безумным спросом на них. Нет никаких сомнений, что «Оземпик» (Ozempic, семаглутид) и «Вегови» (Wegovy, семаглутид), GLP1RA-препараты «Ново Нордиск», и «Мунджаро» (Mounjaro, тирзепатид) и «Зепбаунд» (Zepbound, тирзепатид), GLP1RA/GIPRA-препараты «Илай Лилли», еще долгое время будут лидировать, зарабатывая десятки миллиардов долларов.
Трехлетний курс лечения тирзепатидом против лишнего веса на 94% снизил риск развития сахарного диабета 2-го типа.
ПРЯМАЯ РЕЧЬ
«Ожирение — это не просто косметическая проблема, а серьезное недомогание, ассоциированное с диабетом, болезнями сердца и почек, раком».
Ману Чакраварти (Manu Chakravarthy), старший вице-президент и руководитель глобального подразделения разработки продуктов в области сердечно-сосудистых заболеваний, болезней почек и метаболических расстройств «Рош» (Roche), а до этого — глава исследований и разработок и медицинский директор «Кармот терапьютикс» (Carmot Therapeutics).
«Мы гордимся своей лекарственной программой по борьбе с ожирением и диабетом и уверены, что она, благодаря различным способам применения и комбинирования, удовлетворит потребности и изменит жизнь пациентов к лучшему».
Хезер Тернер (Heather Turner), исполнительный директор «Кармот терапьютикс» (Carmot Therapeutics).
«Эпидемия ожирения — мировой кризис, который продолжает усугубляться, обещая к 2035 году охватить почти половину населения планеты — более 4 миллионов человек. Это создает невероятную нагрузку на общество и системы здравоохранения по всей Земле».
Тим Куцки (Tim Kutzkey), председатель совета директоров «Кармот терапьютикс» (Carmot Therapeutics).
«Собранные клинические данные вселяют оптимизм и дают надежду вывести на рынок лучшую в своем классе терапию, предоставляющую стойкое снижение веса и надежный контроль над уровнем глюкозы».
Леви Гаррауэй (Levi Garraway), исполнительный вице-президент, медицинский директор и руководитель глобального развития продукции «Рош» (Roche).
«Ожирение — гетерогенное заболевание, которое способствует развитию многих других болезней, и всё это налагает значительное бремя на весь мир».
Томас Шинекер (Thomas Schinecker), исполнительный директор «Рош» (Roche).
СУТЬ ВОПРОСА
Современные пептидные GLP1RA-препараты разработаны с прицелом на пролонгированную фармакокинетику так, чтобы их терапевтическое назначение осуществлялось редко — один раз в неделю. Для реализации этого пришлось прибегнуть к модификации аминокислотной последовательности для устойчивости к протеолитическим ферментам, конъюгации с жирными кислотами для обратимого связывания с альбумином и предотвращения клубочковой фильтрации, ковалентному связыванию с фрагментами антител [1].
Пептидные компоненты всех одобренных GLP1RA-препаратов берут начало либо от самого глюкагоноподобного пептида-1 (GLP1), либо от его паралога эксендина-4 (exendin-4). Оба родоначальника являются агонистами GLP1 (GLP1A) с низкой наномолярной аффинностью и высокой потентностью к выработке циклического аденозинмонофосфата (цАМФ) [2].
Сохранение всех этих фармакологических свойств в контексте структурных модификаций, необходимых для улучшения фармакокинетики, было основным принципом изучения взаимосвязи структуры и активности для лираглутида (liraglutide) и семаглутида (semaglutide) [3].
Однако более широкое признание сложности сигнализации рецепторов, сопряженных с G-белком (GPCR), заставило пересмотреть этот подход. Растет интерес к идее создания более эффективных и более переносимых GLP1A за счет тонкой настройки взаимодействия с нижележащими сигнальными путями, что обычно называют смещенным агонизмом [4].
Сейчас большего всего новых лекарств придумывают биотехнологические стартапы, но почти все деньги уходят в карман «Большой фармы».
КАК ЭТО РАБОТАЕТ
«Кармот», воспользовавшаяся фирменной платформой поиска лекарственных соединений «Эволюция хемотипов» (Chemotype Evolution), провела последовательные итерационные этапы синтеза и скрининга библиотек в целях эффективной оптимизации молекул-кандидатов с должным набором свойств и характеристик, включая сигнализацию, биодоступность, долю активного вещества препарата, период полувыведения. И вот что у нее получилось.
Когда инкретиновый гормон в лице GLP1 или глюкозозависимого инсулинотропного полипептида (GIP) связывается со своим рецептором, относящимся к GPCR, этот рецептор меняет форму, что приводит к активации комплекса G-белка и нижележащего сигнального пути цАМФ. Попутно происходит рекрутинг бета-аррестина к рецептору, что отражается снижением сигнализации G-белка (частично за счет транспортировки рецептора). Если ослабить или устранить процесс рекрутинга бета-аррестина, можно добиться более мощного терапевтического эффекта в виде более сильного и продолжительного снижения уровня глюкозы и потери веса [1].
Для этого «Кармот» обратилась к смещенному агонизму рецепторов GLP1 и GIP, спроектировав свои молекулы так, чтобы они активировали цАМФ при минимальном или нулевом рекрутинге бета-аррестина. Избавление от рекрутинга и интернализации (эндоцитоза) бета-аррестина, которые ведут к десенсибилизации (деградации) рецепторов, результировало расширившимся терапевтическим окном с продолжительной фармакологической активностью и улучшенной переносимостью.
Тирзепатид (tirzepatide), предложенный «Илай Лилли» как «Мунджаро» и «Зепбаунд», — на сегодня единственный одобренный двойной агонист рецепторов GLP1 или GIP. Он обладает смещенным агонизмом только в отношении GLP1 с очень низким рекрутингом бета-аррестина, но не делает этого в отношении GIP.
Как полагает «Кармот», ее экспериментальные препараты способны оказать больший терапевтический эффект, чем тирзепатид, если говорить о степени контроля над гликемией и снижении массы тела, и характеризуются менее выраженными нежелательными явлениями.
Еженедельные инъекции тирзепатида — минус 24 килограмма за полтора года.
ПОЧЕМУ ЭТО ВАЖНО
Современные GLP1RA-препараты совершили революцию в лечении ожирения: прежде небывалая эффективность снижения веса, по сути не требующая каких-либо решительных мер по изменению образа жизни, заставила миллионы людей вновь поверить в себя.
Однако применение этих лекарственных средств столкнулось с серьезным дозозависимым ограничением: при увеличении назначаемой дозы растет частота нежелательных явлений (НЯ) со стороны желудочно-кишечного тракта (ЖКТ). Вот почему многие не могут воспользоваться всей мощью GLP1RA-препаратов ввиду невозможности выйти к высокой дозе из-за непереносимости таких НЯ, как тошнота, рвота, диарея, запор. Более того, в целях снижения частоты НЯ приходится следовать продолжительному (вплоть до 20 недель) курсу постепенного повышения дозы, чтобы организм адаптировался к GLP1RA-препаратам. Наконец, инъекционные лекарства требуют особых условий хранения при пониженной температуре.
«Ребелсас» (Rybelsus, семаглутид) производства «Ново Нордиск», единственный на рынке пероральный GLP1RA-препарат, обязывает принимать себя хотя бы за 30 минут до еды: ввиду низкой биодоступности лекарства существует риск снижения терапевтического эффекта.
Novo Nordisk выпустила семаглутид в виде таблеток.
ПАЙПЛАЙН
CT-388 (RG6640) — пептидно-низкомолекулярный GLP1RA/GIPRA, назначаемый подкожными инъекциями один раз в неделю для терапии ожирения и сахарного диабета 2-го типа (СД2).
CT-868 (RG6641) — пептидно-низкомолекулярный GLP1RA/GIPRA, применяемый подкожными инъекциями один раз в день в качестве дополнения к инсулинотерапии сахарного диабета 1-го типа при сопутствующем лишнем весе или ожирении.
CT-996 (RG6652) — пероральный низкомолекулярный GLP1RA, используемый один раз в день для терапии ожирения и СД2. Не исключено, препарат найдет применение для поддерживающего режима после прохождения курса по снижению веса, инициированного инъекционными лекарствами.
КЛИНИЧЕСКИЕ ПОДРОБНОСТИ
CT-388
Клиническое исследование NCT04838405 фазы I изучило еженедельные подкожные инъекции CT-388 (8 или 22 мг) среди взрослых пациентов (n=46), страдающих либо ожирением или избыточной массой тела, либо ожирением и сахарным диабетом 2-го типа (СД2).
По прошествии 24 недель назначение CT-388 в дозе 22 мг привело к снижению веса на усредненных 18,8% (95% ДИ [здесь и далее]: −23,6, −14,0) среди лиц с ожирением (индекс массы тела [ИМТ] ≥ 30,0 кг/м2) и без СД2 относительно плацебо (p<0,001): −18,9±1,1% против −0,1±2,1% [1] [2].
Был отмечен дозозависимый эффект: по истечении 12 недель получавшие 8- или 22-мг дозу препарата потеряли абсолютных 10,2±1,3% и 12,4±0,9% веса, или 9,3% (−13,4, −5,2) и 11,5% (−15,1, −7,9) относительно плацебо.
Похудение минимум на 5%, 10%, 15% и 20% было зарегистрировано для соответственно 100%, 85%, 70% и 45% участников, получавших 22-мг дозу CT-388.
Зафиксировано снижение уровня (p<0,001) гликированного гемоглобина (HbA1c) и глюкозы натощак: на 0,4% и 10,2 мг/дл относительно плацебо. Все испытуемые, у которых был преддиабет до начала исследования, вышли к нормогликемии.
Профиль безопасности CT-388 беспокойств не вызвал: спектр и частота нежелательных явлений, носивших в основном легко-умеренную степень выраженности, укладывались в привычные для препаратов инкретинового класса рамки.
CT-868
Клиническое исследование NCT05110846 фазы II проверило ежедневные подкожные инъекции CT-868 (1,75; 3,25 или 4 мг) среди взрослых пациентов (n=103) с СД2 (HbA1c 7–10%) и сопутствующим ожирением или лишним весом.
По прошествии 26 недель назначения CT-868 уровень HbA1c снизился на 1,16%, 1,63% и 2,03% — против его роста на 0,30% в группе плацебо. Уровень глюкозы в крови удалось довести до целевого при СД2 (HbA1c ≤ 6,5%) среди 52%, 72% и 69% участников — против 18%. К нормогликемии (HbA1c < 5,7%) вышли 13%, 28% и 24% испытуемых — против 5%. Уровень глюкозы натощак по отношению к плацебо снизился на 53–60 мг/дл [1].
Улучшился липидный профиль: за счет падения уровней общего холестерина, холестерина липопротеинов низкой плотности (ЛПНП), холестерина липопротеинов очень низкой плотности (ЛПОНП), триглицеридов, аполипопротеина B.
Применение CT-868 обеспечило уменьшение массы тела на 2,3%, 3,0% и 5,7% — против ее снижения на 2,3% в группе плацебо.
CT-996
Продолжается клиническое испытание NCT05814107 фазы I, тестирующее ежедневное пероральное назначение CT-996 (в разных дозах) среди взрослых пациентов (n=95) с ожирением или лишним весом, наличием СД2 или без него.
В части 2 исследования участники (n=25) с ожирением и без СД2 были разнесены на три когорты, следовавших следующим схемам приема CT-996 или плацебо:
Ступенчатое повышение дозы препарата осуществлялось быстро (на 4-й, 8-й, 15-й и 22-й день), чтобы без промедлений зарегистрировать какие-либо ранее упущенные или неожиданные сигналы, касающиеся безопасности.
После 4 недель лечения вес уменьшился на 2,3%, 5,8% и 7,3% в когортах CT-996 — против его снижения на 1,2% в группе плацебо (н/з, p<0,01, p<0,001). В когорте 3 разница с плацебо составила 6,1% [1].
Установлено, что уровень CT-996 в крови почти не зависел от его приема натощак или вместе с пищей с высоким содержанием жира.
Каких-либо серьезных проблем с безопасностью не выявлено. Ввиду стремительного повышения дозы CT-996 нежелательные явления со стороны желудочно-кишечного тракта носили частый характер: в дальнейшем, когда прибавка дозы будет проводиться более плавно, их частота снизится. Однако частота пульса выросла на 15 уд./мин (это много, если сравнивать с другими препаратами), а один человек столкнулся с повышением уровня липазы (свидетельствует о проблемах с поджелудочной железой).
Девять месяцев каждодневного лечения — и минус 15 килограмм лишнего веса.
ЧТО ДАЛЬШЕ
Продолжается 48-недельное клиническое исследование NCT06525935 фазы II (n=450), тестирующее CT-868 при либо ожирении, либо лишнем весе с сопутствующим ему заболеванием (преддиабет, гипертония, дислипидемия, обструктивное апноэ сна, сердечно-сосудистое заболевание).
ЭКСПЕРТНЫЕ КОММЕНТАРИИ
Если сравнивать эффективность CT-388, инъекционного GLP1RA/GIPRA, с коммерциализированным тирзепатидом и экспериментальным VK2735 авторства соответственно «Илай Лилли» и «Вайкинг терапьютикс» (Viking Therapeutics), которые обращаются к аналогичному двойному агонизму этих инкретинов, расклад получается следующим (приведены приблизительные значения, судя по графикам) [1] [2] [3].
За 12 и 24 недели лечения препарат-кандидат «Кармот» (22-мг доза) обеспечил уменьшение массы тела на 12,4% и 18,9% в абсолютном исчислении и на 11,5% и 18,8% в сравнении с плацебо. «Мунджаро» (15-мг доза) это сделал на абсолютных 9,1% и 14,8% и относительных 7,2% и 12,5%. За 12 недель лечения экспериментальный VK2735 (15-мг доза) снизил вес на абсолютных 13,9% и относительных 12,3%.
Таким образом, выходит, тройка инъекционных GLP1RA/GIPRA выстраивается в порядке уменьшения эффективности: VK2735 — CT-388 — тирзепатид.
Необходимо учитывать, что в случае CT-388 и VK2735 выход к целевой терапевтической дозе был осуществлен, согласно протоколам исследований, намного быстрее, чем при применении тирзепатида: начиная с соответственно 8-й и 10-й недель лечения — против 20-й.
Впрочем, справедливости ради, подобные сравнения методологически неверны ввиду различий в протоколах исследований и популяциях пациентов.
Что касается CT-996, перорального GLP1RA, обеспеченная им за 4 недели лечения разница с плацебо в потере веса на 6,1% (абсолютных 7,3%), выглядит конкурентноспособной, если учитывать, что пероральный семаглутид (50-мг доза) «Ново Нордиск» уменьшил массу тела на относительных 12,7% (абсолютных 15,1%) за 68 недель; пероральный орфорглипрон (orforglipron) [45-мг доза], разрабатываемый «Илай Лилли», снизил вес на относительных 10,6% и 12,4% (абсолютных 12,6% и 14,7%) за 26 и 36 недель; пероральный GSBR-1290 (120-мг доза), за которым стоит «Стракча терапьютикс» (Structure Therapeutics), привел к потере относительных 6,9% (абсолютных 6,4%) лишнего веса за 12 недель. Наконец, разрабатываемая «Вайкинг» пероральная рецептура VK2735 (40-мг доза), двойного агониста GLP1R/GIPR, определила похудение на уровне 3,3% относительно плацебо (абсолютных 5,3%) за 4 недели [4].
Мощные и безопасные лекарства для похудения — всё только начинается.
БИЗНЕС
«Кармот», основанная в 2008 году, за всё время своей самостоятельной работы получила инвестиционных вливаний на сумму $385 млн [1].
Ноу-хау «Кармот» завязано на фирменной платформе поиска лекарств Chemotype Evolution, которая была лицензирована у «Сунисис фармасьютикалс» (Sunesis Pharmaceuticals) в феврале 2010 года [2], а затем усовершенствована.
Большинство фармацевтических компаний ищут новые лекарства, осуществляя высокопроизводительный скрининг (HTS) библиотек из миллионов химических соединений. Поскольку эти соединения получены в результате предыдущих попыток поиска лекарств, они могут не содержать идеальных соединений для новых, более сложных задач.
Chemotype Evolution обращается к библиотеке синтетических фрагментов, собираемых вместе и проходящих итерационный процесс улучшения и оптимизации фармакологических характеристик. Получаемые низкомолекулярные или пептидные соединения и их гибриды способны таргетировать мишени, ранее считавшиеся недосягаемыми.
Процесс начинается с дизайна якорной молекулы, или «приманки», полученной из известных ингибиторов, субстратов, кофакторов, пептидов или хитов, отобранных в результате скрининга фрагментов. Затем «приманка» дериватизируется с помощью линкерного мотива, связывающего ее в индивидуальном порядке с каждым фрагментом из обширной коллекции «Кармот». В результате формируется библиотека из огромного числа гибридов «приманки» и фрагментов. Далее каждый гибрид проходит биофизическую или биохимическую проверку на связывание с мишенью. Наконец, из любых гибридов можно собрать любые другие гибриды, если это представляется релевантным и оптимальным.
Особую силу Chemotype Evolution придает возможность итераций: в отличие от традиционного HTS, когда статическая библиотека проверяется один раз против определенной мишени, в каждом цикле Chemotype Evolution создается новая библиотека.
В начале января 2014 года «Кармот» заключила партнерское соглашение с «Амджен» (Amgen) на предмет использования Chemotype Evolution [3] [4]. Итогом стал соторасиб (sotorasib), предназначенный для лечения местнораспространенного или метастатического немелкоклеточного рака легкого (НМРЛ) с мутацией KRASG12C, которая, как фармакологическая мишень, на протяжении четырех десятилетий считалась недостижимой. В конце мая 2021 года Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) и в начале января 2022 года Европейское агентство по лекарственным средствам (EMA) одобрили соответственно «Лумакрас» (Lumakras, соторасиб) и «Лумикрас» (Lumykras, соторасиб).
Соторасиб — таргетный препарат для лечения местнораспространенного или метастатического немелкоклеточного рака легкого с мутацией KRAS G12C.
В начале января 2023 года «Кармот» выделила Chemotype Evolution в самостоятельное предприятие — «Кимиа терапьютикс» (Kimia Therapeutics), которое пробует найти новые молекулы для лечения онкологических, иммунологических и воспалительных заболеваний [5].
Активы «Кармот» нужны «Рош» по нескольким причинам. Во-первых, гонка фармпроизводителей в стезе борьбы с ожирением затронула всех игроков отрасли, и было бы неправильным отказываться от соперничества, пусть даже напряженного и тяжелого, ведь это направление бизнеса сулит огромные барыши.
Во-вторых, наработки «Кармот» могут быть скомбинированы с экспериментами «Рош», касающимися похудения без потери мышечной массы — следующего этапа конкуренции, активно прорабатываемого в индустрии. В руках швейцарской фармкомпании есть экспериментальный RG6237 (RO7204239, GYM329) — разработанное японской «Шугай фармасьютикал» (Chugai Pharmaceutical) антитело, элиминирующее латентный миостатин из плазмы и тканей в целях сохранения и укрепления мышц.
В-третьих, «Рош» важно диверсифицировать и обновлять свой лекарственный портфель. В 2023 году предприятие заработало 60,44 млрд швейцарских франков (67,16 млрд долларов), из которых ₣44,61 млрд ($49,57 млрд) пришлись на продажи фармацевтической продукции, а ₣14,10 млрд ($15,67 млрд) — диагностической. Почти половина (43%) доходов от торговли лекарствами поступила со стороны онкологических препаратов, а треть (33%) денег принесли лекарства против неврологических и иммунологических расстройств. Всё бы ничего, но патентная защита рано или поздно падет, а продолжать наращивать обороты надо.
ТОЛСТАЯ НЕДАЛЬНОВИДНОСТЬ
В 2018 году, за три года до того, как «Вегови» вышел из стен «Ново Нордиск», став первым современным и мощным препаратом для лечения ожирения, «Рош» отказалась от сильной таблетки для похудения. Фармкомпания из Базеля не воспользовалась своим «правом преимущественного приобретения» экспериментального OWL833 для терапии СД2, разработанного «Шугай», с которой «Рош» дружит вот уже два десятилетия.
Семаглутид поможет сбросить 15% лишнего веса за год. И даже больше.
Непептидный низкомолекулярный препарат-кандидат OWL833 был готов к началу клинических испытаний фазы I и оценивался лишь в несколько десятков миллионов долларов. В том же году его приобрела «Илай Лилли» (Eli Lilly) за авансовых $50 млн [1] — и в итоге он стал называться орфорглипроном (orforglipron, LY3502970).
Этот пероральный GLP1RA, успешно проходящий клиническую проверку в лечении ожирения и СД2, ожидается к регуляторному одобрению к 2026 году. Согласно отраслевым оценкам, у орфорглипрона есть потенциал заработать $50 млрд за шесть лет своей коммерческой доступности, а в 2032 году он доберется до отметки в $14,4 млрд годовых продаж.
Решение «Илай Лилли» поставить на орфорглипрон и профинансировать его доведение до ума наглядно демонстрирует, что крупнейшие игроки на рынке лечения СД2 располагают большим преимуществом в понимании особенностей метаболизма и потери веса, и потому способны принимать верные решения, пусть даже долгосрочные в своей конечной реализации.
В 2018 году научные знания о GLP-1 были относительно небогатыми, а ажиотажа вокруг снижения веса не было и в помине, равно как плотного изучения инкретинов для коррекции иных расстройств за пределами ожирения — так что расставленные «Рош» приоритеты вполне объяснимы.
Опять же, после неудавшихся в 2010 году позднестадийных испытаний таспоглутида (taspoglutide), экспериментального инъекционного еженедельного GLP1RA, лицензированного у французской «Ипсен» (Ipsen), — ввиду серьезных реакций гиперчувствительности и побочных ЖКТ-эффектов — «Рош» сократила инвестиции в диабет и ожирение, сосредоточившись на развитии противоракового портфеля [2] [3] [4].
Недальновидность свойственна всем. Так, «Пфайзер» (Pfizer) в конце 2022 года бесплатно передала «Телевант» (Televant) в составе «Ройвант сайенсиз» (Roivant Sciences) частичную лицензию на RVT-3101 (PF-06480605) — полностью человеческое моноклональное антитело против TNF-подобного лиганда 1A (TL1A), изучаемое в лечении воспалительных заболеваний кишечника. А уже через год «Рош» купила «Телевант» за $7,1 млрд [5].
«Стракча терапьютикс» (Structure Therapeutics) разработала эффективное и безопасное пероральное лекарство, предназначенное для лечения ожирения.
ОСНОВНЫЕ ФАКТЫ
Препарат-кандидат GSBR-1290 относится к агонистам рецептора глюкагоноподобного пептида 1 (GLP1RA) — хорошо изученному классу лекарственных средств, широко применяемых в целях улучшения гликемического контроля при СД2 и долгосрочной коррекции избыточной массы тела при ожирении и лишнем весе.
Основная отличительная черта GSBR-1290 состоит в том, что он сделан в пероральной рецептуре и не нуждается в каких-либо особых требованиях к алгоритму применения: не требуется выдерживать определенное время до приема приема пищи во избежание снижения биодоступности — как это происходит с пероральным семаглутидом (semaglutide).
Клинический путь разработки GSBR-1290 предстоит долгий, но, судя по имеющимся результатам, готовый к реализации препарат получится эффективным и безопасным.
Благодаря своей низкомолекулярной структуре GSBR-1290 несложен для коммерческого производства — соответственно сможет удовлетворить бесконечно усиливающийся спрос на GLP1RA-препараты в условиях их вечного дефицита.
Еженедельные инъекции тирзепатида — минус 24 килограмма за полтора года.
ПРЯМАЯ РЕЧЬ
«К 2030 году число людей, страдающих ожирением, достигнет, как ожидается, одного миллиарда. Вот почему существует высокая потребность в пероральных препаратах, которые легче производить в промышленных масштабах. Они более стабильны, чем инъекционные, что упрощает их транспортировку и хранение. Они более экономичны».
Аня Ястребофф (Ania Jastreboff), директор Йельского центра исследований ожирения (Y-Weight), соруководитель Йельского центра контроля над весом, медицинский директор Йельского центра стресса (Ньй-Хейвен, шт. Коннектикут, США).
«Собранные результаты свидетельствует о значительном потенциале GSBR-1290 стать лучшим в своем классе пероральным низкомолекулярным GLP1RA-препаратом и идеальной основой для будущих комбинированных препаратов, предназначенных для лечения ожирения и сопутствующих заболеваний. Большое окно безопасности позволит в дальнейшем изучить более высокие дозы GSBR-1290».
Реймонд Стивенс (Raymond Stevens), основатель и исполнительный директор «Стракча терапьютикс» (Structure Therapeutics).
КЛИНИЧЕСКИЕ ПОДРОБНОСТИ
Клиническое исследование NCT05762471 фазы Ib/IIa (рандомизированное, двойное слепое, плацебо-контролируемое, многоцентровое) пригласило взрослых пациентов с ожирением или избыточной массой тела.
ФАЗА IB
В фазе Ib, относящейся к исследованию с многократно нарастающими дозами (MAD), пациенты (n=24) были в целом здоровыми и не страдали СД2.
Участники в трех когортах получали перорально плацебо или GSBR-1290 в дозе 30, 60 или 90 мг — один раз в день на протяжении 4 недель.
Назначение GSBR-1290 привело к статистически значимому расхождению с плацебо в том, что касается снижения массы тела: в 60- и 90-мг дозовых когортах уменьшение веса составило абсолютных 5,2% (p=0,002) и 5,4% (p=0,013) от исходного, что эквивалентно потере 4,9 и 4,8 кг [1].
Применение GSBR-1290 отразилось благоприятным изменением ряда кардиометаболических показателей, включая уровень глюкозы в плазме натощак.
GSBR-1290 характеризовался приемлемыми профилями переносимости и безопасности. Никто из пациентов досрочно не прекратил участие в исследовании ввиду нежелательных явлений (НЯ). Среди наиболее распространенных НЯ: тошнота, рвота, диарея, боль в животе, запор, головная боль. Другими словами, в основном зафиксированы НЯ со стороны желудочно-кишечного тракта (ЖКТ), что традиционно для всех GLP1RA-препаратов. Все НЯ носили легко-умеренную степень выраженности.
ФАЗА IIA
Фаза IIa изучила GSBR-1290 на протяжении 12-недельного курса терапии в двух когортах: среди пациентов (n=54) с ожирением или избыточным весом на фоне СД2 (когорта 1) и пациентов (n=64) только с ожирением или избыточным весом (когорта 2).
Среди основных требований к участникам в когорте 1: индекс массы тела (ИМТ) от 27,0 до 40 кг/м2, уровень гликированного гемоглобина (HbA1c) от 7,0% до 10,5%, стабильная доза метформина (≥ 500 мг). Требования в когорте 2: такой же диапазон ИМТ, уровень HbA1c ≤ 6,5%.
Испытуемые ежедневно получали либо плацебо, либо GSBR-1290 в низкой дозе (45 мг) или высокой дозе (90 мг) — когорта 1, в дозе 120 мг — когорта 2. Основной курс лечения осуществлялся после 4-недельного ступенчатого периода повышения дозы до целевой.
12-недельные результаты в когорте 1 пациентов с СД2 получились следующими: [1]
уровень HbA1c снизился на 0,79% и 0,84% в группах 45- и 90-мг дозы GSBR-1290 — против его повышения на 0,18% в группе плацебо (p=0,008 и p=0,001);
вес снизился на 3,3% (3,1 кг) и 3,2% (2,9 кг) — против его прибавки на 0,04% (0,04 кг) (p=0,0019 и p=0,0013];
уровень глюкозы в плазме натощак снизился на 1,61 и 1,28 ммоль/л — против роста на 1,21 ммоль/л (p=0,01 и p=0,0008).
GSBR-1290 обеспечил улучшение уровня постпрандиальной глюкозы, уровня инсулина и резистентности к нему.
В когорте 2 после 12 недель терапии вес снизился на 6,2% (95% ДИ [здесь и далее]: 3,6–8,9) — против отсутствия изменений массы тела (0%) в группе плацебо (p<0,0001). При этом две трети пациентов (67%) потеряли как минимум 6% веса, а одна треть (33%) — не менее чем 10% [2].
Наиболее распространенными НЯ в ответ на назначение GSBR-1290 были таковые со стороны ЖКТ: тошнота, рвота, диарея, снижение аппетита, диспепсия, запор — все они носили легко-умеренную степень выраженности и были преходящими, саморазрешавшимися по ходу лечения.
ТАБЛЕТКИ ПРОТИВ КАПСУЛ
Связующее 12-недельное исследование NCT06139055 фазы I протестировало ежедневную таблетированную рецептуру GSBR-1290 среди взрослых (n=54) с ожирением или лишним весом — до сего момента экспериментальный препарат изучался в капсульной рецептуре.
Назначение GSBR-1290 в 120-мг дозе привело к похудению на 6,9% (4,3–9,4) и 6,8% (4,2–9,4) относительно плацебо — в когортах 1 и 2, различающихся схемами постепенного повышения дозы. Применение GSBR-1290 в 60-мг дозе в когорте 3 обеспечило снижение веса на относительных 6,2% (3,7–8,8) [1].
ЧТО ДАЛЬШЕ
На четвертый квартал 2024 года запланировано 36-недельное клиническое испытание фазы IIb (рандомизированное, двойное слепое, плацебо-контролируемое, многоцентровое, международное), которое проверит таблетированный GSBR-1290 среди взрослых (n=300) с ожирением.
Если всё пойдет удачно, в 2026 году будет организована опорная клиническая программа фазы III с международными исследованиями продолжительностью не менее чем 52 недели; они лягут в основу регистрационного досье.
ПАЙПЛАЙН
Помимо GSBR-1290, на экспериментальном конвейере «Стракча» зреют дополнительные лекарственные программы, ориентированные на лечение ожирения. Все они представлены синтетическими пероральными низкомолекулярными решениями со смещенной активностью, избавленной от ненужной в данном случае бета-аррестиновой сигнализации. Это отражается их усиленной терапевтической эффективностью.
Так, в четвертом квартале 2024 года будет выбран агонист рецептора амилина (AMYR), который поступит в клиническую проверку во второй половине 2025-го.
В первой половине 2025 года «Стракча» определится с агонистом рецептора глюкозозависимого инсулинотропного полипептида (GIPR): его клинические испытания начнутся в первой половине 2026-го.
Тогда же фармпредприятие отберет агонист рецептора глюкагона (GCGR).
Особняком стоит ANPA-0073 — агонист рецептора апелина (APLNR, APJR, APJ), успешно прошедший клиническое исследование фазы Ib. Он во-первых, должен помочь справиться с потерей мышечной массы (за счет наращивания и укрепления мышечной ткани), с которой сталкиваются все худеющие, и, во-вторых, может пригодиться в лечении легочных заболеваний, таких как идиопатический легочный фиброз и легочная артериальная гипертензия (благодаря сглаживанию ремоделирования легочных сосудов, снижению давления в легочной артерии, предотвращения гипертрофии миокарда) [1] [2] [3].
Разумеется, «Стракча» изучает возможность развития комбинированных препаратов: к примеру, сочетающих агонизм GLP1R и GIPR, GLP1R и AMYR, GLP1R и GCGR, а также GLP1R, GIPR и GCGR.
ЭКСПЕРТНЫЕ КОММЕНТАРИИ
На рынке представлен приличный ассортимент GLP1RA-препаратов, но только один из них, «Ребелсас» (Rybelsus, семаглутид) авторства «Ново Нордиск» (Novo Nordisk), реализован в пероральной рецептуре — все прочие сделаны в виде подкожных инъекций. Вот на этот сегмент и нацелен бизнес «Стракча»: считается, что пациентам удобнее пить таблетки, нежели делать уколы, которые к тому же требуют холодовой цепи для длительного хранения.
Novo Nordisk выпустила семаглутид в виде таблеток.
Это не совсем так. Согласно опросу (n=600), три четверти пациентов (77%) с СД2 действительно вначале выказывали свое предпочтение ежедневным пероральным сахароснижающим лекарствам, однако после показа им видеоинструкции, демонстрирующей способ применения еженедельного инъекционного препарата, их отношение изменилось: таблетки теперь выбрала только половина (53%) респондентов [1].
Та степень, с которой GSBR-1290 обеспечил снижение веса, выглядит весьма недурственной и конкурентоспособной (на абсолютных 6,2–6,4% за 12 недель лечения), если исходить из имеющихся на сегодня клинических результатов других пероральных GLP1RA-препаратов. Рассмотрим их в абсолютном выражении, то есть без привязки к группе плацебо.
Так, пероральный семаглутид, назначаемый в 50-мг дозе на протяжении 68 недель в ходе клинического испытания OASIS 1 (NCT05035095) фазы III, привел к похудению на 15,1–17,4% (15,9–18,3 кг).
Одна таблетка в день — минус 18 кг за полтора года.
Орфорглипрон (orforglipron, LY3502970), которым занимается «Илай Лилли», обеспечил в клиническом испытании NCT05051579 фазы II снижение веса на 8,6–12,6% (9,0–13,3 кг) за 26 недель. После 36-недельной терапии масса тела уменьшилась на 9,4–14,7% (9,8–15,4 кг).
Девять месяцев каждодневного лечения — и минус 15 килограмм лишнего веса.
«Рош» (Roche), благодаря приобретенной «Кармот терапьютикс» (Carmot Therapeutics), продвигает CT-996, который привел к похудению на 7,3% за 4 недели в клиническом исследовании NCT05814107 фазы I.
К чему изобретать своё, если можно взять готовое? А ты, Eli Lilly, страшись!
Результаты применения GSBR-1290 при СД2 несколько отстают от исходов в ходе 12-недельного назначения орфорглипрона. Если GSBR-1290 продемонстрировал плацебо-скорректированные снижение уровня гликированного гемоглобина на 1,01–1,02% и уменьшение веса на 3,3–3,5%, то орфорглипрон сделал это на соответствующих 1,02–1,37% и 4,6–7,1%. При этом, однако, эффективность GSBR-1290 приблизительно соответствует таковой у продвигаемого «Ново Нордиск»инъекционного «Вегови» (Wegovy, семаглутид), определившего указанные снижения на 1,0–1,6% и 2,8–4,2%.
В ходе 12-недельного лечения ожирения у недиабетиков терапевтические показатели GSBR-1290 почти совпали с обеспеченными орфорглипроном, хотя всё же несколько уступая препарату «Илай Лилли».
GSBR-1290 характеризовался приемлемой переносимостью: во всяком случае только 5% участников (n=2/37) прекратили терапию из-за НЯ со стороны ЖКТ — против 16-процентной (n=5/31) частоты остановки лечения орфорглипроном.
Как бы то ни было, на данном этапе развития всех этих лекарственных проектов преждевременно делать какое-то итоговое заключение: необходимы долгосрочные результаты крупных клинических испытаний, которые окончательно установят и позволят сравнить эффективность и безопасность пероральных GLP1RA-конкурентов.
БИЗНЕС
«Стракча терапьютикс» (Structure Therapeutics), в начале февраля 2023 года вышедшая на фондовый рынок, настроена серьезно: в конце сентября она, наряду с объявлением о первых успехах GSBR-1290, сообщила о привлечении $300 млн частного финансирования [1] [2] [3].
Хороший старт GSBR-1290 привел к тому, что Уолл-стрит отреагировал ростом биржевых котировок «Стракча» в конце октября того же года.
Однако затем, когда в середине декабря 2023 года были раскрыты дополнительные клинические результаты, недовольный рынок обрушил курс акций «Стракча» на 40%: эффективность GSBR-1290 не смогла значительно опередить обеспечиваемую конкурирующими пероральными GLP1RA-препаратами, одобренными и экспериментальными [4].
В начале июня 2024 года, когда поступили данные об эффективности GSBR-1290 в борьбе с ожирением, ценные бумаги предприятия поднялись на 65% [5]. А затем, впрочем, планомерно скатывались вниз.
В целом «Стракча» представляется сильно недооцененной.
«Стракча», основанная в 2016 году как «Шаути» (ShouTi) и в июне 2022 года получившая нынешнее название, за всё время своей деловой активности смогла собрать $1,1 млрд инвестиционного капитала [6].
Интересным представляется тот факт, что «Стракча» разнесла портфель своей интеллектуальной собственности на три дочерних компании, «Гашербрум байо» (Gasherbrum Bio), «Аннапурна байо» (Annapurna Bio) и «Лхотзе байо» (Lhotse Bio). Это свидетельствует о рисках патентных разбирательств [7].
«Сайтокинетикс» (Cytokinetics) разработала афикамтен (aficamten) — новый лекарственный препарат, предназначенный для лечения симптоматической обструктивной гипертрофической кардиомиопатии.
ОСНОВНЫЕ ФАКТЫ
Гипертрофическая кардиомиопатия (ГКМП) — одно из самых распространенных наследственных заболеваний сердца, вызывающее одышку при нагрузке и снижение физической работоспособности, что ухудшает качество жизни.
Афикамтен, пероральный ингибитор сердечного миозина, успешно прошел итоговую клиническую проверку: он улучшил способность к повседневной физической деятельности и облегчил симптомы обструктивной ГКМП.
В текущем 2024 году «Сайтокинетикс» отправит регистрационное досье афикамтена в адрес регуляторов. На рынок препарат поступит в 2025 году.
Прямым конкурентом афикамтена является «Камзайос» (Camzyos, мавакамтен), в конце апреля 2022 года предложенный «Бристол-Майерс Сквибб» (Bristol-Myers Squibb) и ставший первым в мире препаратом для специфического лечения симптоматической обструктивной ГКМП.
Несмотря на то что афикамтен характеризуется рядом преимуществ перед мавакамтеном (mavacamten), будущий спрос на него упирается в ценовую политику, которую изберет «Сайтокинетикс».
Мавакамтен, разработанный Bristol-Myers Squibb, — новый вздох для всех больных обструктивной гипертрофической кардиомиопатией.
ПРЯМАЯ РЕЧЬ
«Ингибирование сердечного миозина — отличный вариант терапии для пациентов с симптоматической обструктивной гипертрофической кардиомиопатией. Афикамтен, клинически значимым образом улучшающий способность к физической нагрузке, причем без событий с низкой фракцией выброса левого желудочка, когда приходится прерывать лечение, станет желанным дополнением для пациентов и врачей».
Мартин Мэрон (Martin Maron), ведущий автор исследования, директор Центра гипертрофической кардиомиопатии при Больнице и медицинском центре Лэхи (LHMC, Берлингтон, шт. Массачусетс, США).
«Мы убедились, что терапевтический эффект афикамтена охватывает диапазон, связанный с полноценной повседневной активностью. Афикамтен, положительно влияя на сердечную функцию, повышает общую работоспособность при физических нагрузках, что тесно коррелирует с улучшением нескольких важных показателей заболевания, включая структуру и функцию сердца, симптоматику».
Грегори Льюис (Gregory Lewis), соавтор исследования, заведующий кафедрой и секцией сердечной недостаточности, директор лаборатории тестирования сердечно-легочной деятельности, медицинский директор программы трансплантации сердца Центральной больницы Массачусетса (Бостон, шт. Массачусетс, США).
«Фармакологические свойства, заложенные в афикамтене, похоже, воплотились в клинические преимущества, которые до завершения исследования можно было только прогнозировать. Применение афикамтена характеризовалось быстрым выходом к максимальному терапевтическому эффекту у большинства пациентов, а частота нежелательных явлений была схожа с плацебо. Афикамтен — многообещающее лекарственное средство, востребованное при обструктивной гипертрофической кардиомиопатии».
Кэролайн Коутс (Caroline Coats), соавтор исследования, ведущий клиницист Службы наследственных заболеваний сердца Западной Шотландии (Глазго, Великобритания).
«Результаты исследования полностью оправдали высокие ожидания в отношении эффективности и безопасности, продемонстрировав, что афикамтен, добавленный к стандартной терапии, оказал положительное влияние на физическую работоспособность, а также быстрое и устойчивое воздействие на симптомы и функциональный класс у пациентов с обструктивной гипертрофической кардиомиопатии при сохранении безопасности и переносимости».
Фади Малик (Fady Malik), исполнительный вице-президент разработок и исследований «Сайтокинетикс» (Cytokinetics).
«Афикамтен всецело подтвердил свою способность благотворного влияния на качество жизни при обструктивной гипертрофической кардиомиопатии. В случае низкой левожелудочковой фракции нет никаких проблем с тем, чтобы снизить дозу препарата, тем самым индивидуализировав его назначение в целях нивелирования рисков».
Стюарт Купфер (Stuart Kupfer), старший вице-президент, медицинский директор «Сайтокинетикс» (Cytokinetics).
СУТЬ ВОПРОСА
Гипертрофическая кардиомиопатия (ГКМП) — одно из самых распространенных наследственных сердечно-сосудистых заболеваний во всём мире, которым страдает приблизительно каждый второй на тысячу человек [1]. ГКМП, в равной степени поражающая мужчин и женщин, — гетерогенное заболевание с выраженным разнообразием клинических проявлений, естественного течения болезни и ее прогноза [2] [3] [4] [5] [6] [7] [8].
Гипертрофическая кардиомиопатия характеризуется утолщенным, недилатированным (нерасширенным) левым желудочком [2] [4] [7], часто вызывает одышку при нагрузке и снижение физической работоспособности, тем самым ухудшая качество жизни [4] [7] [9] [10].
У приблизительно 70% пациентов с фенотипической ГКМП выявляется обструкция выходного тракта левого желудочка (ВТЛЖ) [15]. Механизмы развития обструкции определены и включают в себя сложное взаимодействие во время систолы между изменениями желудочкового кровотока и асимметричной гипертрофией межжелудочковой перегородки и створками митрального клапана. Именно обструкция является одним из основных факторов, определяющих связанные с ГКМП осложнения, и поэтому выступает важной целью лечения [11] [12] [13].
Гиперконтрактильность (усиленная сократимость) сердца, которая возникает из-за чрезмерного количества актин-миозиновых поперечных мостиков в саркомерах миокарда, является ключевым механизмом, способствующим обструкции оттока. Другие факторы включают удлинение створок митрального клапана, апикальное смещение папиллярных мышц и выпячивание гипертрофированной межжелудочковой перегородки в ВТЛЖ [12] [14]. Препятствие кровотоку создает градиент давления в ВТЛЖ, который может быть надежно определен с помощью эхокардиографии [2] [4] [7] [11] [13].
ПОЧЕМУ ЭТО ВАЖНО
Лечение обструктивной гипертрофической кардиомиопатии (ГКМП) осуществляется как медикаментозно, так и инвазивно.
Фармакологические препараты для лечения обструктивной ГКМП представлены бета-блокаторами, блокаторами кальциевых каналов, дизопирамидом (disopyramide). Однако их эффективность ограничена, включая субоптимальное снижение градиента давления в выходном тракте левого желудочка (ВТЛЖ) и недостаточное облегчение симптомов. Не доказано, что данные лекарственные средства улучшают объективные показатели физической нагрузки. Их назначение приводит к определенным побочным эффектам, недопустимым у некоторых пациентов [1] [2] [3] [4] [5] [6].
Инвазивные методы лечения обструктивной ГКМП, показанные лицам с выраженными симптомами или функциональным дефицитом, такие как хирургическая миоэктомия и чрескожная спиртовая абляция межжелудочковой перегородки, эффективны для уменьшения градиента давления в ВТЛЖ и благоприятно влияют на клиническое течение, включая длительное облегчение ограничивающих физическую активность симптомов [7]. Однако эти вмешательства связаны с риском и обычно проводятся только в отдельных крупных хирургических центрах [8].
Современные стратегии лечения обструктивной ГКМП привели к тому, что большинство пациентов достигают продолжительности жизни, нормальной или близкой к ней, а показатели заболеваемости улучшились. Тем не менее по-прежнему высока потребность в новых лекарственных препаратах, которые были бы безопасными и надежно улучшали самочувствие и функционирование пациентов с минимальными побочными эффектами [2] [5].
КАК ЭТО РАБОТАЕТ
Гипертрофическая кардиомиопатия (ГКМП) возникает в результате патогенных генетических мутаций, затрагивающих гены, кодирующие белки сердечного саркомера, такие как миозин [1] [2]. Такие мутации, есть мнение, механистически увеличивают чистое производство энергии в саркомере in vitro [3] [4] [5] [6]. Результаты исследований согласуются с основной патофизиологией миокарда левого желудочка у пациентов с ГКМП: он характеризуется гиперконтрактильностью (сильным сокращением) со сниженным комплаенсом (растяжимостью) [7]. Всё это расширило понимание молекулярного патогенеза ГКМП и стимулировало усилия по поиску модуляторов сердечного миозина (моторного белка, управляющего сокращениями сердечной мышцы), способных воздействовать на основной механизм гиперконтрактильности при обструктивной гипертрофической кардиомиопатии [8].
Афикамтен (aficamten, CK-3773274, CK-274), разработанный «Сайтокинетикс» (Cytokinetics), представляет собой низкомолекулярный селективный обратимый аллостерический ингибитор сердечного миозина нового поколения. Афикамтен, посредством ингибирования активированной актином АТФазы в миозине с одной головкой и миозине с двумя головками, стабилизирует миозин в освобожденном состоянии после эффективного удара, делая невозможной гидролизацию АТФ [9] [10].
Афикамтен уменьшает количество активных актин-миозиновых поперечных мостиков во время каждого сердечного цикла, тем самым подавляя гиперконтрактильность миокарда, лежащей в основе патофизиологии гипертрофической кардиомиопатии. Предполагаемый фармакологический эффект афикамтена заключен в снижении силы, создаваемой саркомерами, что отражается уменьшением обструкции выходного тракта левого желудочка (ВТЛЖ) и улучшением диастолической функции. Снижение сократительной силы сердца должно облегчить обструкцию ВТЛЖ за счет противодействия чрезмерному утолщению левожелудочковой стенки в этом тракте [11] [12] [13].
Прямое ингибирование сердечного миозина для снятия гиперконтрактильности и обструкции ВТЛЖ при обструктивной ГКМП — это таргетный фармакологический подход, направленный на улучшение функциональных возможностей пациентов и симптомов их заболевания.
Весной 2022 года «Бристол-Майерс Сквибб» (Bristol-Myers Squibb) предложила первое такое лекарство в лице препарата «Камзайос» (Camzyos, мавакамтен), который улучшает способность к физической нагрузке и облегчает симптомы у пациентов с обструктивной ГКМП. Мавакамтен (mavacamten) является низкомолекулярным селективным аллостерическим ингибитором сердечной миозиновой АТФазы [14] [15] [16] [17].
Несмотря на схожий механизм действия, фармакокинетический профиль афикамтена несколько отличается от такового у мавакамтена. Так, период полувыведения мавакамтена составляет 7–9 дней, а равновесное состояние в плазме достигается приблизительно через 6 недель [14] [18]. У афикамтена существенно более короткий период полувыведения (где-то 3,4 дня), а на выход к равновесному состоянию уходит 2 недели, что позволяет быстрее подбирать его терапевтическую дозу и, в случае необходимости, быстрее отменять его действие путем снижения дозы [11].
КЛИНИЧЕСКИЕ ПОДРОБНОСТИ
Опорное клиническое исследование SEQUOIA-HCM (NCT05186818) фазы III (рандомизированное, двойное слепое, плацебо-контролируемое, многоцентровое, международное) пригласило взрослых пациентов (n=282) с симптоматической гипертрофической кардиомиопатией и обструкцией выходного тракта левого желудочка.
Среди основных требований к участникам:
диагноз гипертрофической кардиомиопатии, подтвержденный левожелудочковой гипертрофией без расширения (дилатации) камеры левого желудочка при отсутствии другого кардиологического заболевания, с конечно-диастолической толщиной стенки левого желудочка либо ≥ 15 мм в как минимум одном миокардиальном сегменте, либо ≥ 13 мм в как минимум одном сегменте стенки и с известной генетической мутацией, вызывающей гипертрофическую кардиомиопатию, или семейным анамнезом последней;
пиковый градиент давления выходного тракта левого желудочка (LVOT-G) в покое ≥ 30 мм рт. ст. и LVOT-G после пробы Вальсальвы ≥ 50 мм рт. ст.;
фракция выброса левого желудочка (LVEF) ≥ 60%;
хроническая сердечная недостаточность II или III функционального класса по NYHA;
На протяжении 24 недель испытуемые ежедневно перорально получали плацебо или афикамтен (в дозе 5, 10, 15 или 20 мг). Разрешался фоновый прием уже назначенных кардиологических препаратов, таких как бета-блокаторы, верапамил, дилтиазем, дизопирамид.
Первичная конечная точка эффективности лечения обструктивной гипертрофической кардиомиопатии была установлена изменением пикового потребления кислорода (pVO2), согласно кардиопульмональному нагрузочному тесту (CPET).
Применение афикамтена привело к росту этого показателя на усредненных 1,8 (95% ДИ [здесь и далее]: 1,2–2,3) мл/кг/мин — против изменения на 0,0 (−0,5, 0,5) мл/кг/мин в группе плацебо. Расхождение получилось статистически значимым: среднеквадратичная средняя разница составила 1,7 (1,0–2,4) мл/кг/мин (p<0,001) [1].
Афикамтен обеспечил статистически значимые (p<0,0001) и клинически значимые улучшения всех десяти вторичных конечных точек, включая:
суммарный балл клинических симптомов по Канзасскому опроснику для больных кардиомиопатией (KCCQ-CSS) после 12 и 24 недель терапии (W12 и W24): +11 в группе афикамтена — против +5 в группе плацебо;
снижение функционального NYHA-класса хотя бы на один (W12 и W24): у 49% и 59% пациентов — против 18% и 24%;
LVOT-G после выполнения физических упражнений (W12 и W24): −44,8 и −47,6 мм рт. ст. — против +2,8 и +1,8 мм рт. ст.;
пропорцию пациентов с LVOT-G < 30 мм рт. ст. (W12 и W24): 52% и 49% — 6% и 4%;
продолжительность статуса пригодности к септальной редукционной терапии на протяжении 24-недельного периода лечения: +37 дней — против +114 дней;
изменение общей рабочей нагрузки, согласно CPET на 24-й неделе терапии: + 14 Вт — против +1 Вт.
Уровень N-концевого фрагмента мозгового натрийуретического пептида (NT-proBNP) после 24 недель лечения изменился на среднегеометрических пропорциональных +0,20 (0,17–0,22) пг/мл — против +1,00 (0,91–1,07) пг/мл.
Афикамтен характеризовался приемлемой переносимостью: профиль нежелательных явлений (НЯ) был сравним с таковым у плацебо. Серьезные НЯ, вызванные лечением, были зарегистрированы у 5,6% пациентов в группе афикамтена — против 9,3% в группе плацебо.
ЧТО ЕЩЕ
Согласно промежуточным 48-недельным результатам непрерывного назначения афикамтена в FOREST-HCM (NCT04848506) обеспечило следующие терапевтически благотворные изменения в ходе лечения обструктивной гипертрофической кардиомиопатии (n=46) [1]:
LVOT-G в покое −39,6 мм рт. ст.;
LVOT-G по Вальсальве −53,2 мм рт. ст.;
улучшение функционального NYHA-класса на ≥1 у 82%;
снижение уровня NT-proBNP на 63%;
улучшение показателей структуры и функции сердца, включая уменьшение максимальной толщины стенок, снижение индекса объема левого предсердия, уменьшение соотношения E/e′;
снижение на 94% риска пригодности для септальной редукционной терапии.
Безопасность афикамтена не вызывала каких-либо претензий. Наблюдалось умеренной снижение левожелудочковой фракции выброса (−5,1 мг). Ввиду ее уменьшения ниже 50% трем пациентам потребовалась коррекция дозы афикамтена: у двух всё протекало бессимптомно, у третьего случился рецидив фибрилляции предсердий из-за алкоголя.
ЧТО ДАЛЬШЕ
В рамках клинической программы афикамтена «Сайтокинетикс» оценивает его потенциал как лекарственного средства, улучшающего способность к физической нагрузке и облегчающего симптомы у пациентов с гипертрофической кардиомиопатией, а также его долгосрочное влияние на структуру и функцию сердца. Для этого осуществляются следующие клинические испытания фазы II/III и III:
MAPLE-HCM (NCT05767346): лечение афикамтеном симптоматической обструктивной гипертрофической кардиомиопатии у взрослых — сравнение с метопрололом;
ACACIA-HCM (NCT06081894): лечение афикамтеном симптоматической необструктивной гипертрофической кардиомиопатии у взрослых;
FOREST-HCM (NCT04848506): продолжение назначения афикамтена сроком до 5 лет в целях выяснения его долгосрочных безопасности и эффективности среди участников других испытаний.
ЭКСПЕРТНЫЕ КОММЕНТАРИИ
Обеспеченное афикамтеном улучшение пикового потребления кислорода, согласно кардиопульмональному нагрузочному тесту, оказалось больше в сравнении с результатами применения препарата «Камзайос» (Camzyos, мавакамтен), продвигаемого «Бристол-Майерс Сквибб»: соответствующие разницы с плацебо составили 1,74 (1,04–2,44) мл/кг/мин и 1,4 (0,6–2,1) мл/кг/мин.
Благотворное действие афикамтена проявилось во всех предварительно заданных подгруппах пациентов, включая тех, кто следовал или нет фоновой терапии бета-блокаторами: соответствующие разницы с плацебо составили 1,6 (0,7–2,5) мл/кг/мин и 1,9 (0,8–3,1) мл/кг/мин — это открывает для афикамтена двери для монотерапевтического использования.
Поскольку период полувыведения у афикамтена укорочен по сравнению с таковым у мавакамтена, существуют весомые предпосылки, что подбор адекватной для пациента терапевтической дозы будет происходить быстрее.
Профиль безопасности афикамтена выглядит более приемлемым, нежели таковой у мавакамтена. Так, снижение фракции выброса левого желудочка до менее чем 50% было зарегистрировано у 3,5% пациентов, принимавших афикамтен (притом что никто не прекратил лечения по этой причине), — против 6% при назначении мавакамтена.
Более того, инструкция по медицинскому применению лекарственного препарата «Камзайос» снабжена «чернорамочным» предупреждением относительно рисков развития сердечной недостаточности или полной блокады функции желудочков, так как мавакамтен уменьшает систолическое сокращение.
БИЗНЕС
Когда в конце 2023 года «Сайтокинетикс» объявила об успешной итоговой клинической проверке афикамтена, на волне благостных известий ее биржевые котировки прибавили свыше 80%: рыночная капитализация фармацевтического предприятия превысила 8 млрд долларов [1].
Приблизительно тогда же витали слухи, будто игроки «Большой фармы» присматриваются к покупке «Сайтокинетикс»: приобретение якобы было интересно «АстраЗенека» (AstraZeneca), «Новартис» (Novartis) и «Джонсон энд Джонсон» (Johnson & Johnson). Затем, впрочем, это желание якобы пропало [2] [3] [4].
Согласно отраслевым прогнозам, афикамтен, если будет одобрен, способен выйти на уровень ежегодных продаж $3 млрд долларов к 2035 году.
«Камзайос» (Camzyos, мавакамтен), который «Бристол-Майерс Сквибб» получила благодаря поглощению «Майокардиа» (MyoKardia) в ноябре 2020 года за $13,1 млрд наличными [5], заработал в 2022 и 2023 гг. $24 млн и $231 млн. За первую половину 2024-го он принес $223 млн.
С учетом того, что для незастрахованных американских пациентов стоимость годового курса лечения обструктивной гипертрофической кардиомиопатии препаратом «Камзайос» составляет без малого $100 тыс. (без учета скидок и дисконтов) [6], «Сайтокинетикс», возможно, разумно сыграть на большей ценовой доступности афикамтена.
ПАЙПЛАЙН
ГЕННАЯ ТЕРАПИЯ
Гипертрофическая кардиомиопатия (ГКМП) вызывается мутациями в генах, кодирующих белки сократительного аппарата кардиомиоцитов, что приводит к дезорганизации, фиброзу и утолщению миоцитов, в результате чего возникает дисфункция желудочков. Выявлено свыше 450 мутаций в 20 белках, связанных с саркомерами и миофиламентами, с различной фенотипической выраженностью и тяжестью заболевания. Подавляющее большинство мутаций, определяющих развитие ГКМП, происходит либо в гене MYH7, который кодирует тяжелую цепь бета-миозина, либо в гене миозин-связывающего белка C (MYBPC3), что в совокупности составляет приблизительно две трети известных патологических мутаций [1] [2] [3].
На это и рассчитывает «Теная терапьютикс» (Tenaya Therapeutics), разрабатывающая генную терапию TN-201 для лечения взрослых и детей с ГКМП по причине мутаций в гене MYBPC3. Однократно внутривенно вводимый аденоассоциированный генно-терапевтический препарат доставляет в организм функциональную копию MYBPC3 в целях остановки прогрессирования заболевания и обращения вспять его последствий [4] [5] [6] [7] [8].
Запущено клиническое исследование MyPEAK-1 (NCT05836259) фазы Ib, изучающее TN-201 среди взрослых (n=15) с симптоматической необструктивной ГКМП. Первые результаты будут получены ближе к концу 2024 года.
«Ингибиторы сердечного миозина дали новую надежду пациентам, но они не воздействуют на основную причину гипертрофической кардиомиопатии. Однократная генная терапия это делает, причем она работает как при обструктивных, так и необструктивных формах заболевания; в случае последних пока не доказано, что ингибиторы сердечного миозина помогают».
Фараз Али (Faraz Ali), исполнительный директор « терапьютикс» (Tenaya Therapeutics).
МОДУЛЯЦИЯ САРКОМЕРА МИОКАРДА
«Эджвайз терапьютикс» (Edgewise Therapeutics) занимается EDG-7500 — первым в своем классе пероральным низкомолекулярным селективным модулятором саркомера миокарда, предназначенным для замедления скорости раннего сокращения, снижения диастолического напряжения, улучшения диастолического наполнения и устранения нарушений расслабления сердца, связанных с гипертрофической кардиомиопатией и другими заболеваниями диастолической дисфункции. Мишень EDG-7500, который не оказывает прямого ингибирующего воздействия на моторную головку сердечного миозина, пока остается загадкой [1].
Согласно доклиническим данных на животных моделях обструктивной и необструктивной ГКМП, экспериментальный EDG-7500 вызывает благоприятный ответ при низком риске снижения фракции выброса левого желудочка ниже нормы при всех испытанных дозах. EDG-7500 характеризуется самоограничивающимся эффектом на систолическое сокращение, что позволяет применять его в фиксированной дозе без интенсивного мониторинга безопасности пациентов [2] [3] [4] [5] [6] [7] [8].
Продолжается клиническое исследование CIRRUS-HCM (NCT06347159) фазы II среди взрослых (n=55) с обструктивной и необструктивной ГКМП [9].
«Мы предполагаем, что EDG-7500 продемонстрирует мощное снижение градиента давления выходного тракта левого желудочка при сохранении его нормальной сократимости, а также обеспечит улучшение наполнения желудочков и диастолическую функцию».
Марк Семигран (Marc Semigran), директор по развитию «Эджвайз терапьютикс» (Edgewise Therapeutics).
ВОССТАНОВЛЕНИЕ ЭНЕРГЕТИЧЕСКОГО ГОМЕОСТАЗА МИОКАРДА
«Имбрия терапьютикс» (Imbria Pharmaceuticals) успешно провела препарат-кандидат нинерафаксстат (ninerafaxstat, IMB-101, IMB-1018972) через клиническое исследование IMPROVE-HCM (NCT04826185) фазы II среди взрослых (n=67) с симптоматической необструктивной гипертрофической кардиомиопатией и готовится к запуску FORTITUDE-HCM фазы III [1] [2] [3].
Нинерафаксстат — пероральный низкомолекулярный сердечный митотроп, предназначенный для восстановления энергетического гомеостаза миокарда [4] [5] [6]. Основной механизм действия нинерафаксстата заключен в частичном ингибировании окисления жирных кислот путем прямого конкурентного ингибирования тиолазы I (3-кетоацил-коэнзим-А-тиолаза, 3-KAT) — последнего фермента в митохондриальном пути бета-окисления длинноцепочечных жирных кислот [4] [5] [7]. Это приводит к смещению энергетического метаболизма сердца с окисления свободных жирных кислот на окисление глюкозы, уменьшая количество кислорода, необходимого на моль вырабатываемого аденозинтрифосфата (АТФ), и тем самым повышая эффективность работы сердца, особенно при ограниченном поступлении кислорода. Способность нинерафаксстата оптимизировать энергетическую эффективность сердца может привести к улучшению функции при физической нагрузке за счет улучшения расслабления, наполнения и ударного объема миокарда во время нагрузки [6].
«Улучшения, наблюдаемые при использовании нинерафаксстата как в отношении симптомов, так и функциональных возможностей пациентов с необструктивной гипертрофической кардиомиопатией, для которых нет одобренных вариантов лечения, явно поддерживает дальнейшую клиническую оценку этого препарата».
Мартин Мэрон (Martin Maron), ведущий автор исследования, директор Центра гипертрофической кардиомиопатии при Больнице и медицинском центре Лэхи (LHMC, Берлингтон, шт. Массачусетс, США).
ВОССТАНОВЛЕНИЕ ГОМЕОСТАЗА МЕДИ
Академические учреждения, возглавляемые Манчестерским университетом (Великобритания), пробуют, на рельсах клинического испытания TEMPEST (NCT04706429) фазы II, перепрофилировать триентин (trientine), используемый в лечении болезни Вильсона хелатирующий медь II агент, направив его против гипертрофической кардиомиопатии у взрослых (n=154) [1] [2]
Идея состоит в том, что при ГКМП нарушен гомеостаз меди (повышен уровень меди и церулоплазмина в сыворотке крови), а несвязанные и слабосвязанные тканевые ионы меди II являются мощными катализаторами реактивных форм кислорода (ROS) и окислительного стресса, а также ингибиторами ферментативных антиоксидантов, таких как внеклеточная супероксиддисмутаза (SOD). Триентин, удаляющий ионы меди II из тканей, ассоциирован с восстановлением ультраструктуры митохондрий и нормализацией экспрессии и ферментативной активности белков, участвующих в энергетическом обмене, компонентов митохондриальной дыхательной цепи и ферментов, участвующих в окислении жирных кислот [3] [4] [5] [6] [7] [8] [9] [10] [11].
Это потенциально очень важно в случае ГКМП, поскольку широко распространена гипотеза, что энергетическое истощение является механизмом, посредством которого генные мутации приводят к фенотипу заболевания [12]. У носителей генов с мутациями саркомеров наблюдается значительно ухудшенная энергетика миокарда еще до развития левожелудочковой гипертрофии, то есть, как предполагается, дефицит энергии является первичным событием [13]. Нарушение энергетики миокарда при ГКМП связано с прогрессирующим фиброзом миокарда [14]. Более того, наследственные дефекты в производстве энергии митохондриями и окислении жирных кислот приводят к фенотипам, имитирующим ГКМП [15]. Триентин также нормализует внеклеточную SOD, что подавляет ROS-опосредованную активацию тканевого фактора роста бета (TGF-β) и обращает фиброз миокарда [4] [5] [6].
Ввиду определенных проблемы с безопасностью хелаторов тяжелых металлов, включая нейротоксичность и гепатотоксичность, некоторые склоняются к использованию природных хелатирующих агентов, например куркумина [16].
«Айкерво» (Iqirvo, элафибранор) — новый лекарственный препарат, предназначенный для лечения взрослых пациентов с первичным билиарным холангитом (ПБХ).
Первичный билиарный холангит (ПБХ) — редкое прогрессирующее аутоиммунное заболевание печени, ранее называвшееся первичным билиарным циррозом, характеризуется скоплением жёлчи и токсинов (холестаз) и хроническим воспалением, которые вызывают необратимый фиброз (рубцевание) печени и разрушение жёлчных протоков.
Элафибранор (elafibranor) — пероральный низкомолекулярный двойной агонист альфа- и дельта-рецепторов, активируемых пероксисомными пролифераторами (PPARα и PPARδ) [1].
«Айкерво» назначается в сочетании с урсодезоксихолевой кислотой (ursodeoxycholic acid, UDCA) при недостаточном терапевтическом ответе на нее или самостоятельно в случае непереносимости последней.
«Айкерво» одобрен Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) в начале июня 2024 года [2]. Регуляторный вердикт вынесен в условном порядке, то есть лекарственному средству предстоит подтвердить свою клиническую пользу, продемонстрировав, к примеру, продление жизни или предотвращение печеночной декомпенсации.
Элафибранор разработан французской «Дженфит» (Genfit), которая в середине декабря 2021 года передала французской «Ирсен» (Ipsen) права на коммерциализацию препарата за авансовых €120 млн, последующие выплаты до €360 млн по мере его реализации и двузначное роялти от продаж [3].
Для американских пациентов месячный курс лечения первичного билиарного холангита при помощи «Айкерво» обойдется в $11,5 тыс.
Согласно отраслевым прогнозам, пиковые продажи элафибранора достигнут $500 млн в год.
В клиническом исследовании ELATIVE (NCT04526665) фазы III (рандомизированном, двойном слепом, плацебо-контролируемом, многоцентровом, международном), охватившем взрослых пациентов (n=161) с ПБХ, ежедневное назначение 80 мг элафибранора на фоне UDCA на протяжении 52 недель обеспечило выход к биохимическому ответу для 51% испытуемых (n=55/108) — против 4% (n=2/53) в группе плацебо и UDCA (p<0,001) [4].
Под биохимическим ответом, как композитной конечной точкой, понималось сочетание следующих лабораторных показателей: снижение уровня сывороточной щелочной фосфатазы (СЩФ) ниже предела с максимумом в 1,67 раза выше верхней границы нормы (ВГН), не менее чем 15-процентное падение уровня СЩФ, снижение уровня общего билирубина хотя бы до ВГН.
Нормализация уровня СЩФ, как показателя прогрессирования поражения печени, начала отмечаться уже после 4 недель лечения и была зафиксирована для 15% пациентов (n=16/108) — против 0%(n=0/53) [p=0,002].
Применение элафибранора не привело к статистически значимому (p=0,20) ослаблению кожного зуда среди пациентов, страдающих умеренно-тяжелым таковым (балл ≥ 4): снижение на 1,93 балла по числовой рейтинговой шкале наихудшего зуда (WI-NRS) — против снижения на 1,15 балла.
Однако пациенты всё равно сообщили о заметном подъеме качества жизни, прежде нарушенном ввиду обременительности кожного зуда. Улучшения касались выраженности зуда, продолжительности и непрерывности сна, эмоционального воздействия зуда [5] [6].
Среди наиболее распространенных нежелательных явлений (НЯ) в ответ на назначение элафибранора: увеличение массы тела, абдоминальная боль, диарея, тошнота, рвота.
Продолжительные наблюдения установили, что по прошествии 78 недель лечения ПБХ элафибранором устойчивый биохимический ответ был зарегистрирован для 70% пациентов (n=19/27) против 0% (n=0/13), а нормализация СЩФ — для 19% (n=5/27) против 0% (n=0/13) [5] [7].
В середине августа 2024 года FDA одобрило «Ливделзи» (Livdelzi, селаделпар) — пероральный низкомолекулярный селективный агонист дельта-рецептора, активируемого пероксисомными пролифераторами (PPARδ). Селаделпар (seladelpar), разработанный «Симабей терапьютикс» (CymaBay Therapeutics), которую купила «Гилеад сайенсиз» (Gilead Sciences), является прямым конкурентом элафибранора.
«Ливделзи» (Livdelzi, селаделпар) — новый лекарственный препарат, предназначенный для лечения первичного билиарного холангита у взрослых.
ОСНОВНЫЕ ФАКТЫ
Первичный билиарный холангит (ПБХ; ранее назывался первичным билиарным циррозом) — редкое прогрессирующее аутоиммунное заболевание печени, приводящее к фиброзу, циррозу и преждевременной смерти.
Не существует способов, позволяющих окончательно вылечить ПБХ.
Нормализация биомаркеров холестаза вкупе с ослаблением выраженности зуда, обеспечиваемые селаделпаром, представляют собой явное изменение в парадигме лечения ПБХ.
Селаделпар назначается в сочетании с урсодезоксихолевой кислотой при недостаточном терапевтическом ответе на нее или самостоятельно в случае непереносимости последней.
Селаделпар не рекомендован к применению при декомпенсированном циррозе (асцит, варикозное кровотечение, печеночная энцефалопатия).
«Ливделзи» одобрен Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) в середине августа 2024 года [1].
Регуляторное решение вынесено в условном порядке: на базе суррогатного биомаркера, представленного снижением уровня щелочной фосфатазы. Препарату предстоит окончательно подтвердить клиническую пользу.
Селаделпар разработан «Симабей терапьютикс» (CymaBay Therapeutics), которую в конце марта 2024 года купила «Гилеад сайенсиз» (Gilead Sciences) за $4,3 млрд [2] [3].
Американским пациентам месячный курс лечения первичного билиарного холангита при помощи «Ливделзи» обойдется в $12,6 тыс., что на 10% и 30% дороже, чем «Айкерво» (Iqirvo, элафибранор) и «Окалива» (Ocaliva, обетихолевая кислота), другие препараты против ПБХ.
ПРЯМАЯ РЕЧЬ
«Диагноз первичного билиарного холангита ставится всё большему числу людей. Пациенты страдают от непрекращающегося зуда или ощущения мурашек по коже, а также изнурительной усталости, которая усугубляется зудом по ночам. Появление селаделпара — важная веха для всего сообщества».
Кэрол Робертс (Carol Roberts), президент PBCers Organization (США), некоммерческой организации поддержки пациентов с ПБХ.
«Селаделпар выводит лечение ПБХ на качественно новый уровень, поскольку существующие препараты, хронически принимаемые для замедления поражения печени и сдерживания прогрессирования, не помогают в 40% случаев. У многих пациентов сохраняются ненормальные печеночные показатели и не исчезает зуд, один из основных симптомов».
Палак Триведи (Palak Trivedi), ученый-клиницист и гепатолог из Бирмингемского университета (Великобритания).
«Селаделпар — несомненный признак медицинского прогресса, знаменующий наступление новой эры в лечении ПБХ, которое одновременно предоставляет положительные биохимические результаты и улучшает качество жизни, что является весьма желательными совпадением целей клиницистов и пациентов».
Дэвид Ассис (David Assis), гепатолог из Йельского медицинского института (Нью-Хейвен, шт. Коннектикут, США).
«Селаделпар — потенциально лучший в своем классе препарат, который изменяет сложившуюся картину течения ПБХ, не только улучшая и даже нормализуя показатели печеночной функции, но и облегчая кожный зуд как особенно тяжелый симптом».
Тимоти Уоткинс (Timothy Watkins), вице-президент по клинической разработке противовоспалительных лекарств «Гилеад сайенсиз» (Gilead Sciences).
«Люди, живущие с ПБХ, много лет ждали каких-либо изменений в стандартах лечения. Регуляторное одобрение многообещающего «„Ливделзи“ открыло новую страницу в медицинских подходах к ведению этой обременяющей болезни».
Дэниел О’Дэй (Daniel O’Day), председатель правления и исполнительный директор «Гилеад сайенсиз» (Gilead Sciences).
СУТЬ ВОПРОСА
Первичный билиарный холангит — редкое прогрессирующее аутоиммунное заболевание печени, при котором негнойный холангит (воспаление жёлчных протоков) по неизвестной причине поражает преимущественно междольковые жёлчные протоки, что приводит к портальной гипертензии и циррозу печени [1] [2].
Первичным билиарным холангитом страдают в основном женщины старше 40 лет [3]. Заболеваемость составляет 0,33–5,8 на 100 тыс. человеко-лет, распространенность — 1,91–40,2 на 100 тыс. человек [4].
Патогенез первичного билиарного холангита изучен не до конца и, по-видимому, включает генетическую предрасположенность и факторы окружающей среды [5] [6] [7].
Холестатическое поражение печени характеризуется различной скоростью иммуноопосредованного разрушения внутрипеченочных жёлчных протоков, сопровождающегося портальным воспалением [8] [9] [10] [11] [12]. Дуктопения (потеря жёлчных протоков) приводит к холестазу (нарушению оттока желчи из печени в двенадцатиперстную кишку) и гепатоцеллюлярному повреждению, прогрессирующему поражению печени с развитием фиброза, конечной стадии болезни печени, печеночной недостаточности [13] [12] [14] [15]. Независимо от стадии заболевания многие люди испытывают значительное ухудшение качества жизни, особенно из-за утомляемости и зуда [2] [8] [14].
Первичный билиарный холангит ассоциирован с другими аутоиммунными состояниями, такими как склеродермия, синдром Шегрена, саркоидоз, аутоиммунный гепатит.
В отличие от других причин цирроза, портальная гипертензия и варикозное расширение вен пищевода могут развиваться до появления цирроза [10] [11].
Гистологическое повреждение, характеризующееся гранулематозным лимфоцитарным холангитом, ассоциировано с ненормальными печеночными пробами, включая повышение уровня сывороточной щелочной фосфатазы (СЩФ), гамма-глутамилтрансферазы (ГГТ), активности аминотрансфераз и общего билирубина [16]. Эти биохимические показатели болезни коррелируют с тяжестью течения первичного билиарного холангита, эффективностью его лечения и исходами [17] [18] [19] [20] [21].
Уровень СЩФ как минимум в 1,67 раза выше верхней границы нормы (ВГН) и уровень общего билирубина выше ВГН служат суррогатными показателями активности заболевания, ассоциированы с риском прогрессирования болезни, с достаточной вероятностью предсказывают клиническую пользу лечения [18] [22]. Продемонстрировано, что исходы улучшаются при нормализации уровней СЩФ и общего билирубина [20].
Первоочередное лечение первичного билиарного холангита обращается к назначению урсодезоксихолевой кислоты с последующим, если биохимический ответ недостаточен, добавлением обетихолевой кислоты или фибратов (например, безафибрата, фенофибрата) [10] [11] [12] [23]. Терапии усталости не предусмотрено, а зуд лечится с переменным успехом [24]. Многим пациентам в конечном итоге может потребоваться трансплантация печени [25] [12] [14].
БОЛЬШИЕ ЧИСЛА
Согласно отраслевым прогнозам, селаделпар выйдет на уровень ежегодного заработка в $650 млн, а пиковых $2,9 млрд достигнет к 2036 году.
Оптимистичные предположения отталкиваются от допущения, что значительная часть пациентов, принимающих препарат «Окалива» (Ocaliva, обетихолевая кислота) авторства «Интерсепт фармасьютикалс» (Intercept Pharmaceuticals), которую в ноябре 2023 года купила итальянская «Альфасигма» (Alfasigma) за $800 млн [1] [2], перейдет на селаделпар или будет использовать его в качестве дополнительного лекарственного средства. Но это непростая задача, ведь обетихолевая кислота (obeticholic acid), агонист фарнезоидного X-рецептора (FXR), в течение ближайших восьми лет лишится патентной защиты, что породит ее недорогие генерические копии.
Тем не менее все предпосылки высокого интереса к селаделпару существуют. Во-первых, неоптимальный ответ на первоочередное назначение урсодезоксихолевой кислоты отмечается у 15–40% пациентов [3] [4], притом что 3–5% сталкиваются с неприемлемыми нежелательными явлениями [4]. Во-вторых, биохимический ответ на применение обетихолевой кислоты регистрируется у менее чем 50% больных [5].
КАК ЭТО РАБОТАЕТ
Селаделпар (seladelpar, MBX-8025) — мощный селективный агонист дельта-рецептора, активируемого пероксисомными пролифераторами (PPARδ), снижающий уровень биохимических маркеров холестаза, повреждения печени и ее воспаления [1] [2] [3].
PPARδ, активируемый жирными кислотами транскрипционный фактор, участвует в метаболизме жирных кислот и воспалении [4] [5] [6]. В печени гены, регулируемые PPARδ, экспрессируются в гепатоцитах, холангиоцитах, клетках Купфера и звёздчатых клетках [7] [8]. PPARδ играет критическую роль в гомеостазе жёлчных кислот и оказывает противофиброзное действие [9] [4] [5] [6] [10].
Агонизм PPARδ ассоциирован со снижением синтеза жёлчных кислот, подавлением воспалительных цитокинов, ингибированием пролиферации и активации звёздчатых клеток.
Оригинатором селаделпара является «Орто-МакНил-Янссен фармасьютикалс» (Ortho-McNeil-Janssen Pharmaceuticals), подразделение «Джонсон энд Джонсон» (Johnson & Johnson), которое в августе 2006 года лицензировало молекулу «Симабей терапьютикс» (CymaBay Therapeutics), на тот момент называвшейся «Метаболекс» (Metabolex) [11].
КЛИНИЧЕСКИЕ ПОДРОБНОСТИ
Клинические исследования ENHANCE (NCT03602560) и RESPONSE (NCT04620733) фазы III (рандомизированные, двойные слепые, плацебо-контролируемые, многоцентровые, международные) охватили взрослых пациентов (n=265 и n=193) с первичным билиарным холангитом и недостаточным ответом на назначение урсодезоксихолевой кислоты или с ее непереносимостью.
Среди основных требований к участникам: уровень сывороточной щелочной фосфатазы (СЩФ) как минимум в 1,67 раза выше верхней границы нормы, несмотря на назначение урсодезоксихолевой кислоты на протяжении хотя бы 12 месяцев.
Испытуемые получали ежедневно перорально плацебо или селаделпар в дозе 5 мг (только в ENHANCE) или 10 мг — на протяжении 3 или 12 месяцев в ENHANCE и RESPONSE соответственно.
Композитная первичная конечная точка эффективности лечения ПБХ была установлена пропорцией пациентов, биохимически ответивших на терапию. Ответ фиксировался при удовлетворении всем следующим критериям: снижение уровня СЩФ ниже предела с максимумом в 1,67 раза выше верхней границы нормы (ВГН), не менее чем 15-процентное падение уровня СЩФ, снижение уровня общего билирубина хотя бы до ВГН.
К указанному показателю эффективности вышли 78% (n=43/55) и 62% (n=79/128) пациентов в группах, получавших 10 мг селаделпара, — против 13% (n=7/56) и 20% (n=13/65) в группах плацебо (p<0,0001) [1] [2] [3].
Антихолестатический эффект селаделпара подтвердился нормализацией уровня СЩФ (не выше верхней границы нормы) у 27% (n=15/55) и 25% больных (n=32/128) — против 0% (n=0/56 и n=0/65) в контрольных группах (p<0,0001) [2].
Снижение уровня СЩФ составило 44% и 42% — против 4% (p<0,0001).
Применение селаделпара привело к значительному улучшению сывороточных биомаркеров повреждения печени и липидного профиля, таких как АЛТ, ГГТ, холестерин ЛПНП, триглицериды.
Назначение селаделпара обеспечило ослабление зуда: среди пациентов с зудом умеренно-тяжелой степени выраженности (балл ≥ 4 по числовой рейтинговой шкале, NRS) его снижение составило 3,1 и 3,2 пункта (после 3 и 6 месяцев лечения в ENHANCE и RESPONSE) — против снижения на 1,6 и 1,7 пункта в группах плацебо (p=0,02 и p<0,005).
Профиль безопасности селаделпара не характеризовался какими-либо серьезными нежелательными явлениями (НЯ), на которые следовало бы обратить особое внимание.
КОНТРАРГУМЕНТЫ
Селаделпару придется напрямую соперничать с «Айкерво» (Iqirvo), элафибранор), в начале августа 2024 года одобренным FDA с подачи французских «Дженфит» (Genfit) и «Ирсен» (Ipsen) при таком же показании для лечения первичного билиарного холангита.
Если сравнивать терапевтическую эффективность селаделпара и элафибранора (elafibranor), двойного агониста PPARα и PPARδ, хотя методологически это не совсем верно, получается, что выход к композитному биохимическому ответу с коррекцией на плацебо был справедлив для соответственно 42% (95% ДИ [здесь и далее]: 28–53) и 47% (32–57) пациентов, а нормализация СЩФ — для 25% (18–33) и 15% (6–23). Выраженность зуда снизилась на абсолютных 3,2 и 1,9 балла.
Зуд является частым симптомом у пациентов с ПБХ и связан с высокой концентрацией в плазме крови веществ, которые выводятся с желчью. При гепатоцеллюлярной недостаточности зуд, как правило, стихает, что позволяет предположить, что холестатическая печень ответственна за выработку пруритогенных веществ [1].
Элафибранор для лечения ПБХ, когда урсодезоксихолевая кислота не справляется.
Почему назначение селаделпара привело к статистически значимому уменьшению зуда, тогда как применение элафибранора этого не обеспечило?
В исследовании селаделпара у 14% пациентов был цирроз печени, а исходная усредненная интенсивность зуда (среди участников с умеренно-тяжелым таковым) составляла 6,1±1,4 балла в группе препарата и 6,6±1,4 балла в группе плацебо, согласно числовой рейтинговой шкале (NRS). В исследовании элафибранора у 43% испытуемых был мостовидный фиброз или цирроз, а изначальная усредненная интенсивность зуда составляла соответственно 3,3±2,8 и 3,2±2,9 балла, согласно числовой рейтинговой шкале наихудшего зуда (WI-NRS). Помимо различий в дизайне исследований уместно задаться вопросом: связано ли отсутствие преимущества в ослаблении зуда при использовании элафибранора с механистическими особенностями препарата или всё же с более высоким процентом пациентов с циррозом?
Что касается внедрения селаделпара и элафибранора в клиническую практику, в дальнейшем уместно рассмотреть возможность их использования в качестве препаратов первой линии, то есть вместо урсодезоксихолевой кислоты, потому что до 40% пациентов к ней рефрактерны [2], а выдаваемая ею частота биохимического ответа (значительное снижение уровня СЩФ на фоне нормализации уровня общего билирубина), который ассоциирован с хорошим долгосрочным прогнозом [3], не сильно отличается от таковой при применении этих новых препаратов.
Не исключено, разумным окажется первоочередное назначение комбинации из урсодезоксихолевой кислоты и селаделпара или элафибранора.
Селаделпар и элафибранор — новые и запатентованные лекарственные соединения, и потому дорогостоящие в приобретении и присутствующие не на любом рынке. Приемлемым решением является добавление фибратов (давно ставших генерическими, и потому доступных по цене) к урсодезоксихолевой кислоте [4] [5]. Это подтверждено рядом исследований и метаанализов, изучивших подобное применение фибратов вне инструкции: агониста PPARα фенофибрата (fenofibrate) [6] [7] [8] [9] и пан-агониста PPAR безафибрата (bezafibrate) [10] [11] [12] [13] [14]. Добавление фибратов также поможет снять зуд [15].
ЧТО ДАЛЬШЕ
Селаделпар изучается в ходе долгосрочной проверки безопасности и эффективности лечения первичного билиарного холангита в рамках пятилетнего клинического испытания ASSURE (NCT03301506) фазы III среди пациентов из других исследований, пожелавших продолжить терапию.
Согласно промежуточному анализу, охватившему данные за 2 года применения селаделпара, 70% участников (n=99/179) вышли к вышеописанной композитной конечной точке эффективности лечения. При этом у 42% человек отмечена нормализация СЩФ [1].
Организовано трехлетнее клиническое испытание AFFIRM (NCT06051617) фазы III среди пациентов с ПБХ и компенсированным циррозом. Проверяется гипотеза, способен ли селаделпар отсрочить время до наступления одного из следующих событий: смерть по любой причине, трансплантация печени, балл по шкале MELD (модель для оценки терминальной стадии заболевания печени) ≥ 15, требующий лечения асцит, госпитализация по причине печеночной декомпенсации (кровотечение в местах варикозного расширения вен пищевода или желудка, печеночная энцефалопатия, спонтанный бактериальный перитонит).
Результаты AFFIRM, которые должны быть представлены в FDA до февраля 2030 года, лягут в основу полноценного одобрения «Ливделзи».
Клиническая проверка показала, что сочетание препаратов «Опдиво» (Opdivo, ниволумаб) и «Ервой» (Yervoy, ипилимумаб), назначаемое в ходе первоочередного лечения неоперабельного рака печени (гепатоцеллюлярной карциномы), превосходит эффективность, обеспечиваемую терапией в лице препарата «Ленвима» (Lenvima, ленватиниб) или «Нексавар» (Nexavar, сорафениб).
ОСНОВНЫЕ ФАКТЫ
«Бристол-Майерс Сквибб» (Bristol-Myers Squibb) продемонстрировала преимущество сочетания из ниволумаба (nivolumab) и ипилимумаба (ipilimumab), блокаторов PD-1 и CTLA-4, над ленватинибом (lenvatinib) или сорафенибом (sorafenib), тирозинкиназными ингибиторами, продвигаемыми соответственно «Эйсай» (Eisai) / «Мерк и Ко» (Merck & Co.) и «Байер» (Bayer), в ходе перволинейной терапии рака печени.
По отношению к препаратам сравнения иммунноонкологический коктейль снизил риск смерти на 21% и снизил риск прогрессирования заболевания или смерти на 13%.
Этого недостаточно, чтобы опередить нынешний стандарт в лице комбинации из «Тецентрика» (Tecentriq, атезолизумаб) и «Авастина» (Avastin, бевацизумаб) — блокатора PD-L1 и ингибитора VEGF авторства «Рош» (Roche).
Однако в абсолютном исчислении продление общей выживаемости оказалось превосходным.
Регистрационное досье отправлено в адрес регуляторов.
ПРЯМАЯ РЕЧЬ
«Медиана общей выживаемости получилась одной из самых длинных, которые мы когда-либо наблюдали в ходе лечения распространенной гепатоцеллюлярной карциномы. Мы уверены, что разработали новый стандарт лечения».
Питер Галле (Peter Galle), клинический гепатолог из Медицинского центра при Майнцском университете (земля Рейнланд-Пфальц, Германия).
«Отмеченная нами частота уменьшения опухоли — одна из самых высоких среди других вариантов лечения рака печени. Высокий уровень ответа на терапию повышает шансы трансформации заболевания из неоперабельного в поддающееся резекции».
Лаура Гофф (Laura Goff), исполнительный медицинский директор Центра ухода за онкологическими пациентами при Онкологическом центре Вандербильта — Инграма (VICC, шт. Теннесси, США).
«Несмотря на продолжающееся развитие фармакологической науки, прогноз для пациентов с гепатоцеллюлярной карциномой по-прежнему остается плохим. Вот почему важно предложить им новые способы лечения, которые, возможно, помогут».
Дана Уолкер (Dana Walker), вице-президент и руководитель глобальной программы по раку желудочно-кишечного тракта и мочеполовой системы «Бристол-Майерс Сквибб» (Bristol-Myers Squibb).
КЛИНИЧЕСКИЕ ПОДРОБНОСТИ
Клиническое исследование CheckMate 9DW (NCT04039607) фазы III (рандомизированное, открытое, с активным контролем, многоцентровое, международное) пригласило взрослых пациентов (n=668) с распространенной гепатоцеллюлярной карциномой, ранее не проходившей системной терапии.
Участникам назначали либо комбинацию из ниволумаба и ипилимумаба, либо ленватиниб или сорафениб (на выбор исследователя) — до момента прогрессирования заболевания или неприемлемой токсичности.
После наблюдений на протяжении медианных 35,2 месяца (26,8–48,9) результаты получились следующими [1] [2] [3].
Общая выживаемость в группе «Опдиво» с «Ервоем» вышла к 23,7 месяца (95% ДИ [здесь и далее]: 18,8–29,4) — против 20,6 месяца (17,5–22,5) в группе «Ленвимы» или «Нексавара». Риск смерти снизился на относительный 21%: отношение риска (hazard ratio, HR) 0,79 (0,65–0,96); p=0,018.
Вероятность остаться в живых на протяжении 24 месяцев составила 49% против 39%, 36 месяцев — 38% против 24%.
Частота общего ответа (ORR) составила 36% (31–42), включая 7% полных ответов (CR), — против 13% (10–17), в том числе 2% CR (p<0,0001).
Медиана длительности ответа (DoR) получилась равной 30,4 месяца (21,2–NE) — против 12,9 месяца (10,2–31,2).
Медиана выживаемости без прогрессирования (PFS) обозначилась на уровне 9,1 месяца (6,6–10,5) — против 9,2 месяца (7,9–11,1). Риск прогрессирования заболевания или смерти снизился на относительных 13%: HR 0,87 (0,72–1,06). Статус PFS в течение 18 месяцев оказался справедливым для 34% пациентов против 18%, 24 месяцев — 28% против 12%.
Назначение иммуноонкологического коктейля привело к снижению риска ухудшения симптомов заболевания на относительных 24%: HR 0,76 (0,62–0,93); p=0,0059.
КОНТРАРГУМЕНТЫ
Использование ленватиниба или сорафениба в качестве контрольной группы — сомнительный выбор «Бристол-Майерс Сквибб». Разумнее было остановиться на более эффективных схемах, представленных либо сочетанием атезолизумаба (atezolizumab) с бевацизумабом (bevacizumab), за которым стоит «Рош», либо дуэтом «Имфинзи» (Imfinzi, дурвалумаб) с «Имджудо» (Imjudo, тремелимумаб) — блокатора PD-L1 с блокатором CTLA-4, продвигаемым «АстраЗенека» (AstraZeneca). Первая схема является предпочтительной, вторая выступает альтернативной в случае противопоказаний к назначению бевацизумаба или его непереносимости.
Согласно нынешним рекомендациям Американского общества клинической онкологии (ASCO), атезолизумаб с бевацизумабом лидирует по эффективности первоочередного лечения гепатоцеллюлярной карциномы, улучшая выживаемость и сдерживая прогрессирование заболевания относительно сорафениба: OS HR 0,66 (0,52–0,85) и PFS HR 0,65 (0,53–0,81) [1] [2].
Применение дурвалумаба (durvalumab) с тремелимумабом (tremelimumab) характеризуется только улучшением выживаемости относительно сорафениба: OS HR 0,78 (0,67–0,92) и PFS HR 0,90 (0,77–1,05) [3] [4].
ЭКСПЕРТНЫЕ КОММЕНТАРИИ
Начиная с марта 2020 года, «Опдиво» с «Ервоем» применяются во второлинейной терапии гепатоцеллюлярной карциномы. Но бизнес «Бристол-Майерс Сквибб» требует расширения охвата пригодных пациентов.
Результаты клинической проверки этого сочетания в перволинейной терапии рака печени получились на первый взгляд идентичными таковым в случае перволинейного использования дурвалумаба с тремелимумабом. Но есть важные отличия.
Во-первых, «Опдиво» с «Ервоеем» обеспечили более продолжительную общую выживаемость с медианой 20,6 месяца — против 16,4 месяца в случае «Имфинзи» с «Имджудо».
Во-вторых, они повысили шансы остаться в живых: до 49% и 38% на протяжении 24 и 36 месяцев — против 41% и 31%.
В-третьих, на лечение ответила большая пропорция пациентов: ORR 36% и CR 7% — против ORR 20% и CR 3%.
В-четвертых, ответ на лечение продолжался дольше: 30,4 месяца — против 22,3 месяца.
Однако справиться с прогрессированием заболевания удалось аналогично плохо.
Осталось разобраться с нежелательными явлениями и их частотой, чтобы понять, насколько новая схема лечения окажется переносимой.
Перспективность ниволумаба с ипилимумабом в контексте первоочередного лечения гепатоцеллюлярной карциномы сомнений не вызывает, ведь долгосрочные 4-летние наблюдения за пациентами, получавшими дурвалумаб с тремелимумабом, установили прилично высокую пропорцию долгожителей, то есть перешагнувших отметку 36-месячной выживаемости: каждый четвертый (25%) — против 15% в группе сорафениба [1].
В идеале было бы правильным напрямую сравнить эти две схемы с «Тецентриком» и «Авастином», но гранды фармотрасли на такое вряд ли пойдут.
ОДНАКО
Списывать сорафениб или ленватиниб со счетов пока рано. Согласно систематическому обзору и метаанализу, сочетание тирозинкиназного ингибитора, блокатора PD-1 и локорегионарной терапии характеризовалось максимальными шансами на конверсию изначально неоперабельной гепатоцеллюлярной карциномы в поддающуюся радикальной резекции, то есть с потенциалом очень длительной выживаемости [1].
Посттравматическое стрессовое расстройство (ПТСР) — серьезное и опасное для жизни нервно-психическое заболевание.
С ПТСР сталкивается большое количество людей.
Все нынешние методы лечения ПТСР характеризуются явно недостаточной эффективностью.
Разработан препарат для лечения ПТСР, который, располагая совершенно новым механизмом действия и не будучи психоактивным соединением, характеризуется высокой эффективностью, превосходящей селективные ингибиторы обратного захвата серотонина (СИОЗС), такие как сертралин (sertraline) и пароксетин (paroxetine).
У экспериментального лекарства нет серьезных побочных эффектов.
ЧТО ПРОИЗОШЛО
Австралийская «Байономикс» (Bionomics) справилась со среднестадийной клинической проверкой экспериментального анксиолитика BNC210, предназначенного для лечения посттравматического стрессового расстройства (ПТСР).
Терапевтическая эффективность BNC210 значительно превзошла таковую, предоставляемую одобренными лекарствами против ПТСР, притом что существующие препараты не обеспечивают должного выхода к ремиссии для большинства пациентов.
На момент анонса, состоявшегося в конце сентября 2023 года, Уолл-стрит отреагировал ростом биржевых котировок «Байономикс» сразу на 400%. Впрочем, впоследствии курс акций предприятия плавно снизился до исходного уровня.
«Байономикс» продолжает подготовку к запуску регистрационной клинической программы. Если BNC210 получит регуляторное одобрение, он станет первым новым за минувшие два десятка лет лекарством для терапии посттравматического стрессового расстройства.
Препарат-кандидат BNC210 представляет собой первый в своем классе отрицательный аллостерический модулятор никотинового ацетилхолинового рецептора α7, разрабатываемый для лечения тревожных, травматических или стрессовых расстройств.
Параллельно «Байономикс» изучает анксиолитик BNC210 в неотложном лечении социального тревожного расстройства (социофобии), и здесь он может стать первым лекарством, не вызывающим седативного эффекта и привыкания.
ПОЧЕМУ ЭТО ВАЖНО
Посттравматическое стрессовое расстройство (ПТСР) — серьезное нервно-психическое заболевание, с которым ежегодно сталкивается большое количество людей: к примеру, в США и Европе распространенность ПТСР в течение жизни составляет 6,4–8,0% [1] [2] [3].
Особенно широко ПТСР распространено среди участников или свидетелей боевых действий. Тем не менее подавляющее большинство (80% пациентов) с этим диагнозом представлено гражданской популяцией, не имеющей никакого отношения к военным конфликтам [12] [14].
ПТСР — серьезное и опасное для жизни патологическое состояние, ассоциированное с повышенной смертностью, кардиометаболической заболеваемостью и риском суицида. ПТСР отрицательно влияет на повседневную жизнь человека, часто приводит к разрыву отношений, депрессии, снижению повседневной активности, ухудшению когнитивных и психосоциальных функций, злоупотреблению психоактивными веществами и высокозатратному использованию медицинских услуг.
Лечение ПТСР особенно сложно в таких нередких случаях, как диссоциативный подтип ПТСР, повторное воздействие травмы, наличие сопутствующих заболеваний, включая аффективные расстройства и расстройства, связанные с употреблением алкоголя и психоактивных веществ [4] [5] [6].
Комплексная природа ПТСР ассоциирована с обострением симптомов, резистентностью к лечению и прекращением терапии [5] [7]. Психотерапия, ориентированная на травму, является золотым стандартом лечения ПТСР, однако у многих людей симптоматика сохраняется, а частота отказа от лечения высока [8] [9] [10]. Фармакотерапия ПТСР, предполагающая прием селективных ингибиторов обратного захвата серотонина (СИОЗС), не обеспечивает должного эффекта у приблизительно 35–47% пациентов [11].
Для решения проблемы индивидуального, общественного и экономического бремени ПТСР необходимы более эффективные терапевтические подходы [12] [13].
ЧТО ВЫЯСНИЛОСЬ
Клиническое исследование ATTUNE (NCT04951076) фазы IIb (рандомизированное, двойное слепое, плацебо-контролируемое, многоцентровое, международное) пригласило взрослых пациентов (n=212) с диагнозом посттравматического стрессового расстройства (ПТСР).
Среди основных требований к участию: балл выраженности симптомов ≥ 30 по шкале клинической оценки ПТСР по DSM-5 (CAPS-5); индексное травматизирующее событие произошло во взрослом возрасте.
Среди базовых характеристик пациентов: женщин 64%; средний возраст 42 года (19–68); средний балл CAPS-5 41,5 (30–59).
Испытание включило людей с обширным спектром причин, послуживших развитию ПТСР, как то: физическое или сексуальное насилие, внезапная насильственная смерть, нападение с применением оружия, участие или пребывание в зоне боевых действий, дорожно-транспортное происшествие, опасное для жизни заболевание или травма, внезапная смерть от несчастного случая, пребывание в плену, пожар или взрыв, причиненные другому человеку серьезные травмы, вред здоровью или смерть, серьезный несчастный случай на работе, дома или во время активного отдыха, тяжелые человеческие страдания, иной нежелательный или дискомфортный сексуальный опыт, иное очень стрессовое событие или переживание.
На протяжении 12 недель пациенты получали перорально плацебо или BNC210 в дозе 900 мг дважды в день.
Назначение BNC210 обеспечило выход к первичной конечной точке эффективности лечения, установленной изменением общего балла тяжести симптомов ПТСР по шкале CAPS-5: усредненная разница с плацебо составила −4,03 пункта (p=0,048) [1] [2].
Статистически значимая разница с плацебо проявилась даже раньше: на 4-й (p=0,015) и 8-й неделях (p=0,014) терапии: соответственно −4,11 и −4,74 пункта.
Применение BNC210 обеспечило улучшение симптомов, особенно сложных для купирования при ПТСР: симптомы интрузии (критерий B) и негативные мысли и эмоции (критерий D). Так, на 4-й, 8-й и 12-й неделях лечения усредненная разница с плацебо балла интрузии составила −0,98 (p=0,046), −1,35 (p=0,035) и −1,33 (p=0,047) пункта, а балла негативных мыслей и эмоций — −2,05 (p=0,005), −1,79 (p=0,036) и −1,79 (p=0,061) пункта.
Назначение BNC210 привело к существенному улучшению вторичных конечных точек. В частности, на 12-й неделе лечения было отмечено облегчение симптомов депрессии и сна, согласно шкале Монтгомери — Осберг для оценки депрессии (MADRS) и индексу тяжести бессонницы (ISI) соответственно: усредненная разница с плацебо составила −3,19 (p=0,040) и −2,19 (p=0,041) пункта.
Наблюдалась положительная динамика в изменении других показателей, в том числе оцениваемых по шкале общего клинического впечатления о тяжести заболевания (CGI-S), по шкале общего впечатления пациента о тяжести заболевания (PGI-S), по шкале Гамильтона для оценки выраженности тревоги (HAM-A), по шкале инвалидизации Шихана (SDS).
Лечение посттравматического стрессового расстройства при помощи BNC210 характеризовалось приемлемыми переносимостью и безопасностью, тем самым указывая на возможность хронического применения этого препарата.
Среди наиболее распространенных нежелательных явлений (НЯ) при назначении BNC210: головная боль (у 17% пациентов против 13% в группе плацебо), тошнота (12% против 8%), усталость (6% против 8%) — в подавляющем большинстве случаев они носили легко-умеренную степень выраженности.
По сравнению с плацебо не зафиксировано избыточных психоактивных НЯ, таких как седация, привыкание и когнитивные нарушения, что выгодно отличает BNC210 от существующего арсенала лекарственных препаратов против ПТСР. Применение BNC210 не вызывало НЯ сексуального характера, таких как снижение либидо и эректильная дисфункция.
Был отмечен рост уровня печеночных ферментов (у 13% пациентов против 2% в группе плацебо), хотя это не было связано с функциональными нарушениями печени (то есть не было ее лекарственного поражения, декомпенсации печеночного заболевания, проявления закона Хая, повышения уровня биллирубина), протекало бессимптомно и в большинстве случаев разрешалось без необходимости в отмене лечения.
Массив научных данных указывает на участие холинергической системы в этиологии и лечении поведения, связанного со страхом. Никотиновые ацетилхолиновые рецепторы (nAChR) — это ионотропные лиганд-связанные рецепторные полипептиды, активируемые ацетилхолином и никотином [2] [3]. Подтип α7 состоит из пяти субъединиц α7, которые в основном гомомерны [4].
В целом NAChR являются неселективными катионными каналами, причем подтип α7 особенно проницаем для кальция [5], а также для натрия [6]. Исследования на крысах выявили роль α7 nAChR в анксиолитическом поведении. Антагонизм α7 nAChR, концентрация которого высока в миндалевидном теле крыс [7], отразился на поведении, связанного со страхом [8].
Селективные ингибиторы обратного захвата серотонина (СИОЗС), как было показано, ингибируют никотиновые ацетилхолиновые рецепторы, то есть СИОЗС, как предполагается, действуют через холинергическую, а не серотонинергическую модуляцию [9]. Хотя это не было продемонстрировано на людях, крысы засвидетельствовали холинергическое торможение активности миндалевидного тела после приема бензодиазепинов [10]. Это оставляет вероятность для того, что лечение тревоги бензодиазепинами действует, не исключено, через холинергическое торможение в дополнение к ГАМК-ергическим эффектам.
На базе доклинических моделей о роли холинергической системы в поведении, связанном со страхом, высказано предположение, что модуляция α7 nAChR может служить анксиолитиком. С этой целью был разработан BNC210.
В доклинических моделях BNC210 хорошо переносился, был безопасен и не вызывал седативного эффекта [11]. BNC210 эффективно работал как анксиолитик в различных поведенческих парадигмах, в том числе превзошел плацебо в тестах на тревожность: темно-светлая камера (для мышей) и приподнятый крестообразный лабиринт (для крыс) [3]. Изучение профиля безопасности и переносимости BNC210 в исследованиях на людях выявило обнадеживающие результаты без седативных эффектов, присущих типичным анксиолитикам [12] [13].
ЧТО ДАЛЬШЕ
«Байономикс» (Bionomics) в четвертом квартале 2024 года, после консультации с Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA), намеревается организовать опорное клиническое испытание фазы III, которое окончательно установит эффективность и безопасность экспериментального BNC210 для лечения посттравматического стрессового расстройства (ПТСР). Результаты будут собраны в 2025–2026 гг.
Ввиду определенного негативного влияния на печеночные ферменты будет также протестирована сниженная доза BNC210.
ПЕРСПЕКТИВЫ
Как утверждает «Байономикс» (Bionomics), эффект лечения и размер эффекта, обеспечиваемые экспериментальным анксиолитиком BNC210 в ходе лечения посттравматического стрессового расстройства (ПТСР), соответственно превосходят и сравнимы с таковыми при применении селективных ингибиторов обратного захвата серотонина (СИОЗС).
Так, BNC210, согласно шкале клинической оценки ПТСР по DSM-5 (CAPS-5), обеспечил снижение общего балла тяжести симптомов на абсолютных 20 пунктов, тогда как сертралин (sertraline) снизил его на на абсолютных 6,8 и 9,8 пункта (в разных клинических испытаниях), а пароксетин (paroxetine) — на 14 и 11 пункта [1].
Размер эффекта при назначении BNC210 составил 0,40; 0,44 и 0,40 — соответственно после 4, 8 и 12 недель лечения [2].
Лечение при помощи BNC210 прекратили 20% испытуемых — против 10% в контрольной группе, что, впрочем, предсказуемо ввиду нестабильного поведения пациентов с ПТСР.
Прямым конкурентом BNC210 выступает экспериментальная MDMA-психотерапия, в двух клинических исследованиях фазы III, MAPP1 (NCT03537014) и MAPP2 (NCT04077437), продемонстрировавшая снижение общего балла CAPS-5 на абсолютных 26,2 и 23,7 пункта, притом что соответствующий размер эффекта (относительно плацебо) составил 0,91 и 0,7 [3] [4].
Для сравнения: размер эффекта при назначении сертралина и пароксетина изрядно уступает обеспечиваемому MDMA-психотерапией, будучи втрое и вдвое меньше — 0,31–0,37 и 0,45–0,56. Кроме того, если назначение этих СИОЗС сопровождалось высокой частотой выбывания пациентов из клинических испытаний (отказ от лечения на уровне 28–39%), то при MDMA-психотерапии пациенты вообще не стремились ее прекратить.
В ходе MDMA-психотерапии на лечение ответили 80–87% пациентов (клинически значимое снижение общего балла тяжести симптомов ПТСР по шкале CAPS-5 минимум на 10 пунктов), потеряли диагноз 67–71% испытуемых (статус респондента с одновременным прекращением соответствия диагностическим критериям ПТСР), к ремиссии вышли 33–46% человек (потеря диагноза и общий балл CAPS-5 менее 11 пунктов).
Несмотря на приличную результативность MDMA-психотерапии в лечении ПТСР, она завязана на применении психоделического (эмпатогенного) вещества, и потому ее распространение в мире ожидается весьма ограниченным ввиду строгости законодательства в отношении психоактивных соединений, пусть даже используемых исключительно в медицинских целях, поскольку они обладают высоким потенциалом злоупотребления.
Для понимания дальнейших перспектив BNC210 в лечении ПТСР, следует обратить внимание на такие важные для врачей и пациентов показатели, как размер эффекта и степень приверженности лечению, а также пропорции ответивших на лечение пациентов, потерявших диагноз и вышедших к ремиссии. Сведения об указанных пропорциях «Байономикс» пока не раскрыла.
Как бы то ни было, получилось, что терапевтическая эффективность анксиолитика BNC210 сравнима с таковой у MDMA-психотерапии, если речь идет о достаточно тяжелом ПТСР (общий балл тяжести симптомов CAPS-5 ≥ 40).
Существующие лекарства для снижения артериального давления недостаточно эффективны, со временем к ним развивается привыкание.
Блокирование выработки ангиотензиногена в печени — решительно новый действенный способ удержания давления в норме.
Предполагается очень редкий режим дозирования грядущих лекарств: один раз в месяц, квартал или даже полгода.
Зилебесиран (zilebesiran) и тонламарсен (tonlamarsen) — быстро, надежно, дорого!
ЧТО ПРОИЗОШЛО
«Алнайлам фармасьютикалс» (Alnylam Pharmaceuticals) и «Айонис фармасьютикалс» (Ionis Pharmaceuticals) разработали новаторские препараты, предназначенные для лечения гипертонии.
Передовое лечение обещает помочь даже в том случае, если артериальное давление никак не поддается снижению имеющимся арсеналом гипотензивных лекарственных средств.
Экспериментальные зилебесиран (zilebesiran) и тонламарсен (tonlamarsen) обращаются к совершенно новому механизму действия, прежде не задействовавшемуся в лечении гипертонии и предполагающему подавление выработки ангиотензиногена в печени.
Существуют весомые предпосылки, что должная терапевтическая эффективность грядущих лекарств сохранится при очень редком режиме дозирования, когда препараты назначаются подкожными инъекциями один раз в три или даже шесть месяцев.
В дальнейшем эти лекарства могут превратиться в еще более удобные пероральные таблетки или капсулы, принимаемые, к примеру, один раз в неделю или месяц.
ПОЧЕМУ ЭТО ВАЖНО
Артериальная гипертензия, или гипертония, — сложное многофакторное заболевание, проявляющееся повышенным артериальным давлением крови.
Клинический диагноз гипертонии, согласно большинству консенсусных руководств, ставится, когда систолическое артериальное давление превышает 140 мм рт. ст., а диастолическое — 90 мм рт. ст. [1] [2] [3]. При этом, однако, Американская кардиологическая ассоциация (AHA) совместно с Американской коллегией кардиологов (ACC) рекомендуют ставить диагноз артериальной гипертензии при соответствующих показателях выше 130 и 80 мм рт. ст. [4].
Гипертонией страдает каждый четвертый мужчина и каждая пятая женщина на планете — всего свыше 1,28 млрд жителей Земли в возрасте 30–79 лет [5].
Гипертония — одна из ведущих причин преждевременной смерти.
Неконтролируемая гипертония, то есть когда ее терапия не проводится надлежащим образом или она вообще остается без какого-либо лечения, увеличивает риск развития или ухудшения сердечно-сосудистых заболеваний и нарушений мозгового кровообращения — ишемической болезни сердца и инсульта, а также способствует прогрессированию хронической болезни почек [6] [7] [8] [9].
Несмотря на наличие эффективных лекарственных препаратов для снижения давления, фармакотерапия гипертонии оставляет желать много лучшего. При этом половине больных, придерживающихся хронического приема гипотензивных лекарственных средств, не удается выйти к целевым показателям артериального давления [10].
Частично это является следствием, во-первых, неспособности лечащего врача начать или интенсифицировать (усилить) медикаментозную терапию и, во-вторых, плохой приверженности пациентов к ежедневному приему пероральных лекарственных препаратов [11] [12] [13].
Ситуация осложняется тем, что даже когда артериальное давление кажется хорошо контролируемым на основе периодических измерений, контроль над гипертонией может оставаться неоптимальным из-за выраженной вариабельности артериального давления в течение суточного цикла и в долгосрочной перспективе [14] [15] [16].
РЕШЕНИЕ ПРОБЛЕМЫ
Хроническая гиперактивность ренин-ангиотензин-альдостероновой системы (РААС) считается одним из основных факторов патогенеза сердечно-сосудистых заболеваний, включая гипертонию, сердечную недостаточность, сердечную гипертрофию, атеросклероз [1] [2].
Ингибирование сигнального пути РААС ингибиторами ангиотензинпревращающего фермента (АПФ) или блокаторами рецепторов ангиотензина II (БРА) — один из наиболее эффективных методов лечения артериальной гипертензии и сердечной недостаточности со сниженной фракцией выброса (HFrEF) [3] [4] [5] [6] [7].
Однако ключевым препятствием для реализации этого подхода в полную силу является то, что он увеличивает риск целевой токсичности, проявляющейся главным образом гиперкалиемией и нарушением функции почек. Другими словами, приходится прибегать к более низким клиническим дозам, что тем самым ограничивает возможность для оптимального ингибирования РААС, которое иначе предоставило бы более выраженные клинические преимущества в отношении снижения артериального давления [8].
Фармакологическое ингибирование нисходящего каскада сигнального пути РААС также приводит к формированию компенсаторных путей в вышележащем каскаде, что еще больше снижает терапевтическую эффективность гипотензивных лекарственных препаратов [9] [10] [11].
Нацеливание на верхний каскад сигнального пути РААС путем сайленсинга (подавления экспресии) вырабатываемого печенью ангиотензиногена (AGT) — принципиально новый подход к сдерживанию активности РААС [12].
Поскольку ангиотензиноген является единственным предшественником всех пептидов ангиотензина, его сайленсинг характеризуется рядом потенциальных преимуществ по сравнению с большинством существующих способов ингибирования РААС [13] [14] [15] [16] [17] [18].
Так, тканеспецифическое подавление выработки ангиотензиногена в печени, никак не затрагивающее ингибирование РААС в почках, позволяет надеяться на улучшенный профиль безопасности, потому что сохраняются почечный гомеостаз и механизм обратной канальцево-клубочковой связи. В итоге снижаются риски роста уровня калия и почечной дисфункции.
К минимуму сведен риск развития компенсаторных механизмов, которые проявляются в ответ на применение АПФ или БРА и которые парадоксальным образом пытаются восстановить уровни ренина, альдостерона и ангиотензина II.
Более полное ингибирование локальной выработки ангиотензина II в сосудистой или сердечной тканях принесет пользу пациентам с резистентной гипертонией или сердечной недостаточностью.
ALNYLAM PHARMACEUTICALS И НОВАТОРСКОЕ ЛЕЧЕНИЕ ГИПЕРТОНИИ
«Алнайлам фармасьютикалс» (Alnylam Pharmaceuticals) разрабатывает экспериментальный препарат зилебесиран (zilebesiran), построенный на терапевтической модальности РНК-интерференции, неоднократно доказавшей собственную медицинскую состоятельность.
Раннестадийная клиническая проверка зилебесирана оказалась весьма успешной, и потому этот препарат-кандидат приглянулся «Рош» (Roche), которая в конце июля 2024 года оформила с «Алнайлам» партнерское соглашение [1].
Согласно договоренностям, швейцарский фармацевтический гигант выдаст «Алнайлам» авансом 310 млн долларов, а затем будет выплачивать дополнительные суммы по мере разработки, регуляторного одобрения и продаж готового препарата — совокупно до 2,8 млрд долларов. Реализация зилебесирана в США будет осуществляться совместно на равных условиях извлечения прибыли, во всех других странах продажей будет заниматься «Рош», отчисляя «Алнайлам» определенное роялти.
ЗИЛЕБЕСИРАН: КАК ОН РАБОТАЕТ
Ренин-ангиотензин-альдостероновая система (РААС) представляет собой сигнальный каскад, который надежно доказал свою определяющую роль в регуляции артериального давление и подавление которого сопровождается хорошо известными гипотензивными эффектами [1] [2] [3] [4] [5] [6] [7].
Вопрос о том, блокировать или не блокировать РААС, давно не является актуальным в клинических сценариях сердечной недостаточности, диабетической нефропатии, хронической болезни почек. Усилия фармотрасли направлены на оптимизацию этой блокады.
[membership level=»2,3″ show_noaccess=»true»]
Зилебесиран (zilebesiran, ALN-AGT) таргетирован против ангиотензиногена (AGT), который является самым ранним предшественником ангиотензина I (проангиотензина), дающего начало ангиотензину II — олигопептидному гормону, который входит в состав РААС и который вызывает сужение сосудов и повышение артериального давления [8].
Зилебесиран построен на базе РНК-интерференции (RNAi) — естественного механизма сайленсинга генов (подавления их экспрессии) на стадии транскрипции, трансляции, деаденилирования или деградации матричной РНК (мРНК) при помощи малой интерферирующей РНК (миРНК). За открытие RNAi была вручена Нобелевская премия в 2006 году [9].
Зилебесиран вмешивается в работу мРНК гена AGT, что отражается его сайленсингом. Ингибирование синтеза ангиотензиногена в печени приводит к устойчивому снижению уровня ангиотензина I и в конечном итоге ангиотензина II. В следствии этого снижается артериальное давление, поскольку ангиотензин II равно как самостоятельно оказывает сосудосуживающий эффект, так и делает это посредством стимуляции синтеза вазопрессина.
Конструктивно зилебесиран состоит из двух частей: химически модифицированной миРНК против AGT и N-ацетилгалактозаминового (GalNAc) лиганда, связывающего асиалогликопротеиновый рецептор 1 (ASGR1) на гепатоцитах в целях таргетной доставки препарата в печень, где эндосомальное накопление миРНК, медленная их утилизация и рециклинг подпитывают нокдаун AGT на протяжении длительного времени.
Зилебесиран обращается к фирменной технологии GalNAc-конъюгатов Enhanced Stabilization Chemistry Plus (ESC+), которая позволяет осуществлять весьма редкое подкожное введение лекарства, вкупе наделяя его высокой селективностью и широким терапевтическим индексом.
[/membership]
ЗИЛЕБЕСИРАН: ВСЁ ПОЛУЧИТСЯ
В пользу того, что технологический подход «Алнайлам фармасьютикалс» (Alnylam Pharmaceuticals) сработает в задаче лечения гипертонии, свидетельствуют поставленные ею на коммерческие рельсы РНК-интерференционные препараты против редких заболеваний:
«Онпаттро» (Onpattro, патисиран) и «Амвуттра» (Amvuttra, вутрисиран): лечение наследственного транстиретинового амилоидоза (hATTR) с полинейропатией (hATTR-PN), ранее известного как семейная амилоидная полинейропатия (FAP) [1] [2].
«Окслумо» (Oxlumo, лумасиран): лечение первичной гипероксалурии 1-го типа (PH1) [4] [5].
Все эти препараты применяются в режиме редкого дозирования. Так, «Онпаттро» требует внутривенных вливаний один раз в три недели, «Гивлаари» необходимо назначать подкожными инъекциями один раз в месяц, а «Окслумо» и «Амвуттра» вводятся подкожно один раз в квартал.
Кроме того, «Новартис» (Novartis) выпустила«Леквио» / «Сибрава» (Leqvio / Sibrava, инклисиран) — РНК-интерференционный препарат, предназначенный для мощного снижения уровня холестерина липопротеинов низкой плотности (ЛПНП) посредством подкожных инъекций один раз в полгода. За оригинальной идеей «Леквио» / «Сибрава» стоит всё та же «Алнайлам».
Две инъекции инклисирана в год — и о высоком холестерине можно забыть.
ЗИЛЕБЕСИРАН: ЭФФЕКТИВНОСТЬ И БЕЗОПАСНОСТЬ
NCT03934307
Клиническое исследование NCT03934307 фазы I (рандомизированное, двойное слепое, плацебо-контролируемое, многоцентровое) привлекло взрослых пациентов с повышенным систолическим давлением (легко-умеренная гипертония: 130–165 мм рт. ст.), не принимающих каких-либо гипотензивных препаратов (либо ранее не лечивших гипертонию, либо отказавшихся от таких лекарств).
[membership level=»2,3″ show_noaccess=»true»]
Участники получили по одной подкожной дозе зилебесирана (zilebesiran) в различных дозах или плацебо.
Результаты в 200-мг подгруппе зилебесирана по прошествии 8 недель следующие [1]:
Снижение сывороточного уровня ангиотензиногена (AGT) более чем на 90% сохранялось в течение 3 месяцев.
Усредненное снижение систолического артериального давления (САД) и диастолического артериального давления (ДАД) составило 11±2 и 8±1 мм рт. ст., причем у некоторых испытуемых максимальное снижение составило 19 и 12 мм рт. ст.
В подгруппах зилебесирана в дозе 100 мг и выше отмечено устойчивое снижение сывороточного уровня AGT более чем на 90%, при этом в 800-мг подгруппе указанное снижение составило 96–98%.
Назначение зилебесирана в дозе 100 мг и выше обеспечило снижение САД не менее чем на 10 мм рт. ст., при этом в 800-мг подгруппе снижение САД превысило 15 мм рт. ст.
После 24 недель продемонстрированы следующие исходы [3]:
На 3-й неделе наблюдений применение зилебесирана в дозе 100 мг и выше привело к снижению сывороточного уровня AGT более чем на 90%, которое сохранялось на протяжении 12 недель, причем в 800-мг подгруппе указанное снижение было устойчивым в течение 24 недель.
На 8-й неделе наблюдений назначение зилебесирана в дозе 200 мг и выше привело к снижению САД не менее чем на 10 мм рт. ст. и продолжило сохранять свое клинически значимое падение вплоть до 24-й недели.
На 24-й неделе наблюдений в 800-мг подгруппе зилебесирана снижение САД превысило 20 мм рт. ст., причем у 6 из 8 пациентов этого удалось добиться без каких-либо дополнительных гипотензивных препаратов.
Зилебесиран в дозе 200 мг и выше обеспечил стабильное и устойчивое снижение САД и ДАД в дневное и ночное время, что свидетельствует о возможности достижения тонического контроля над артериальным давлением в течение длительного периода.
Назначение зилебесирана характеризовалось приемлемой переносимостью, каких-либо серьезных нежелательных явлений (НЯ) в ответ на его назначение не зарегистрировано.
«Алнайлам фармасьютикалс» (Alnylam Pharmaceuticals) дополнительно изучила 800-мг дозу зилебесирана на фоне низкосолевой диеты или попутного ежедневного назначения 300 мг ирбесартана (irbesartan), блокатора рецепторов ангиотензина II (БРА).
Проверка зилебесирана при рационе с низким содержанием соли, рекомендованной гипертоникам, была необходима для выяснения переносимости лечения ввиду истощения объема жидкости в организме по причине потери натрия, вызванного низкосолевой диетой, что способно привести к гипотензивным событиям.
Добавление ирбесартана результировало дополнительным снижением САД и ДАД на 6,4 и 3,2 мм рт. ст., причем без клинически значимых изменений сывороточных уровней креатинина и калия.
Выводы следующие: низкосолевая диета оказывает дополнительный эффект в виде усиленного снижения артериального давления, которое может упасть слишком низко, и потому в задаче предотвращения вероятных гипотензивных событий может пригодиться, напротив, высокосолевая диета.
«Алнайлам» также протестировала зилебесиран среди пациентов с гипертонией и ожирением II или III степени (индекс массы тела [ИМТ] > 35 кг/м2), но без сахарного диабета. Подтверждено выраженное и устойчивое снижение САД с отражающими механизм действия ростом уровня ренина и снижением уровней ангиотензина II и альдостерона.
[/membership]
KARDIA-1
Клиническое исследование KARDIA-1 (NCT04936035) фазы II (рандомизированное, двойное слепое, плацебо-контролируемое, многоцентровое) проверило мононазначение зилебесирана (zilebesiran) среди взрослых пациентов (n=394) с легко-умеренной гипертонией (усредненное дневное систолическое артериальное давление [САД] в пределах 135–160 мм рт. ст.), ранее не лечивших гипертонию либо следующих стабильным курсом гипотензивных препаратов (в любом случае испытуемые не принимали таковые в ходе исследования) [1].
[membership level=»2,3″ show_noaccess=»true»]
На протяжении 6-месячного двойного слепого периода участники получали либо зилебесиран в дозе 150, 300 или 600 мг каждые 6 месяцев или 300 мг каждые 3 месяца, либо плацебо каждые 3 месяца. В последующем 6-месячном открытом периоде пациенты из группы плацебо переводились на зилебесиран.
Изменения артериального давления (мм рт. см.) относительно плацебо фиксировались двумя способами: посредством суточного мониторирования артериального давления (СМАД) и однократного измерения артериального давления сфигмоманометром (ОИАДС).
По истечение 3 месяцев изменение САД, согласно СМАД, составило: −14,1 (95% ДИ [здесь и далее]: −19,2, −9,0), −16,7 (−21,2, −12,3) и −15,7 (−20,8, −10,6) — среди испытуемых, получивших зилебесиран в дозе 150 мг (каждые 6 месяцев), 300 мг (каждые 3 или 6 месяцев) и 600 мг (каждые 6 месяцев) [p<0,0001].
Изменение САД, согласно ОИАДС: −9,6 (−13,8, −5,3), −12,0 (−15,7, −8,3) и −9,1 (−13,4, −4,8) [p<0,0001].
По прошествии 6 месяцев изменение САД, согласно СМАД, получилось равным: −11,1 (−15,8, −6,4), −14,5 (−19,1, −9,9), −14,1 (−18,9, −9,4) и −14,2 (−18,9, −9,5) — среди участников, которым назначили зилебесиран в дозе 150 мг (каждые 6 месяц), 300 мг (каждые 6 месяцев), 300 мг (каждые 3 месяца) и 600 мг (каждые 6 месяцев) [p<0,0001].
Изменение САД, согласно ОИАДС: −7,5 (−12,4, −2,7), −10,5 (−15,3, −5,7), −12,1 (−17,2, −7,1) и −10,2 (−15,1, −5,3) [p=0,0025, p<0,0001].
Пропорции пациентов, ответивших на лечение, — то есть, согласно СМАД, достигших САД < 130 мм рт. ст. и/или его снижения на ≥ 20 мм рт. ст. без дополнительных гипотензивных лекарственных препаратов, — по прошествии 6 месяцев составили 7%, 31%, 51%, 39% и 47% — среди участников, получивших плацебо, зилебесиран в дозе 150 мг (каждые 6 месяц), 300 мг (каждые 6 месяцев), 300 мг (каждые 3 месяца) и 600 мг (каждые 6 месяцев). Соответствующие отношения шансов (odds ratio, OR) выйти к ответу для подгрупп зилебесирана составили 6,8 (2,4–19,4), 19,7 (6,8–56,9), 10,7 (3,8–30,6) и 17,9 (6,2–51,5) [p<0,0001].
Зилебесиран обеспечил непротиворечивое и устойчивое снижение артериального давления на протяжении всего суточного цикла, включая ночное время.
Назначение зилебесирана характеризовалось приемлемой переносимостью. Нежелательные явления (НЯ), связанные с гиперкалиемией (среди 5% пациентов, получавших зилебесиран, — против 1% в группе плацебо), носили легкую степень выраженности, были преходящими и разрешались самостоятельно. Число случаев гипотонии было небольшим (4% против 1%). Не отмечено клинических значимых изменений почечной или печеночной функций. Были зарегистрированы редкие случаи серьезных НЯ (4% против 7%) и тяжелых НЯ (3% против 4%).
Дополнительный анализ установил, что зилебесиран обеспечил должное снижение САД вне зависимости от пола, возраста, расы, а также исходных САД и расчетной скорости клубочковой фильтрации (eGFR). Разве что влияла исходная концентрация ренина в плазме у темнокожих пациентов: если она была низкой, то и снижение САД было не столь выраженным [5].
[/membership]
KARDIA-2
Клиническое исследование KARDIA-2 (NCT05103332) фазы II (рандомизированное, двойное слепое, плацебо-контролируемое, многоцентровое, международное) изучило добавление зилебесирана (zilebesiran) к гипотензивной терапии взрослых пациентов (n=800) с резистентной гипертонией, которая адекватно не контролируется лекарственными препаратами.
[membership level=»2,3″ show_noaccess=»true»]
Среди критериев включения: усредненное дневное систолическое артериальное давление (САД ) в пределах 155–180 мм рт. ст. или 145–180 мм рт. ст. (соответственно при ранее нелеченной гипертонии и леченной одним–двумя препаратами гипертонии), которое должно снизиться до 130–160 мм рт. ст. после вводного периода исследования.
Вводный 4-недельный открытый период, который был необходим для выравнивания исходных показателей САД, предполагал ежедневный однократный прием какого-либо одного гипотензивного лекарства: 2,5 мг диуретика индапамида (indapamide), 40 мг блокатора рецепторов ангиотензина II олмесартана (olmesartan) или 5 мг блокатора кальциевых каналов амлодипина (amlodipine) [1].
В последующем 6-месячном двойном слепом периоде участники однократно получили подкожную инъекцию 600 мг зилебесирана или плацебо — на фоне продолжения ежедневного приема ранее выбранного гипотензивного препарата (по прошествии 3 месяцев могли быть добавлены дополнительные лекарства, если гипертония не купировалась).
Изменения артериального давления (мм рт. см.) относительно плацебо фиксировались двумя способами: посредством суточного мониторирования артериального давления (СМАД) и однократного измерения артериального давления сфигмоманометром (ОИАДС).
По истечение 3 месяцев изменение САД, согласно СМАД, составило: −12,1 (95% ДИ [здесь и далее]: −16,5, −7,6), −9,7 (−12,9, −6,6) и −4,0 (−7,6, −0,3) — среди получивших зилебесиран испытуемых, придерживавшихся ежедневной гипотензивной терапии индапамидом, амлодипином или олмесартаном (p<0,001, p<0,001, p=0,036).
Изменение САД, согласно ОИАДС: −18,5 (−22,8, −14,2), −10,2 (−13,5, −6,9) и −7,0 (−10,4, −3,6) [p<0,001].
По прошествии 6 месяцев изменение САД, согласно СМАД, получилось равным: −11,0 (−14,7, −7,3), −7,9 (−10,6, −5,3) и −1,6 (−4,4, −1,2) [p<0,001, p<0,001, and p=0,26].
Изменение САД, согласно ОИАДС: −13,6 (−16,9, −10,3), −8,6 (−10,9, −6,3) и −4,6 (−6,8, −2,4) [p<0,001].
Пропорции пациентов, ответивших на лечение, — то есть, согласно СМАД, достигших САД < 130 мм рт. ст. и/или его снижения на ≥ 20 мм рт. ст. без дополнительных (к уже назначаемым) гипотензивных лекарственных препаратов, — по прошествии 6 месяцев составили 64%, 40% и 27% — среди участников, получивших зилебесиран, а затем следовавших терапии индапамидом, амлодипином или олмесартантом. В подгруппах плацебо пропорции таковы: 14%, 14% и 17%. Соответствующие отношения шансов (odds ratio, OR) выйти к ответу для подгрупп зилебесирана составили 12,4 (4,6–33,3), 5,1 (2,4–10,6), 1,8 (0,9–3,4) [p<0,001, p<0,001, p=0,077].
Назначение зилебесирана характеризовалось приемлемой переносимостью. Нежелательные явления (НЯ), в том числе лабораторного характера, носили легкую степень выраженности, были преходящими и разрешались самостоятельно. С гипотонией или ортостатической гипотонией столкнулись 0%, 6% и 5% пациентов, получивших зилебесиран, а затем придерживавших терапии индапамидом, амлодипином или олмесартаном, — против 0%, 3% и 3% в подгруппах плацебо.
[/membership]
KARDIA-3
Продолжается клиническое исследование KARDIA-3 (NCT06272487) фазы II (рандомизированное, двойное слепое, плацебо-контролируемое, многоцентровое), которое оценивает оправданность назначения зилебесирана (zilebesiran) в качестве дополнительного препарата среди пациентов (n=390) с высоким сердечно-сосудистым риском и резистентной гипертонией (систолическое артериальное давление 130–170 мм рт. ст.), которая адекватно не контролируется 2–4 классами гипотензивных лекарств с разными механизмами действия.
Среди основных критериев включения (на выбор): сердечно-сосудистое заболевание в анамнезе, высокий сердечно-сосудистый риск или хроническая болезнь почек (расчетная скорость клубочковой фильтрации [eGFR] 30–60 мл/мин/1,73 м2).
Исследование ожидается к завершению весной 2025 года.
ЗИЛЕБЕСИРАН: ОПТИМИСТИЧНЫЕ ПЕРСПЕКТИВЫ
Согласно консенсусному мнению, снижение артериального давления более чем на 5 мм рт. ст. является клинически значимым, и потому терапевтические показатели, обеспеченные зилебесираном (zilebesiran), выглядят весьма убедительными. Прогнозируемый однозначный коммерческий успех препарата обусловлен главным образом схемой его очень редкого дозирования (один раз в квартал или полгода), поскольку приверженность пациентов фармакотерапии гипертонии решительно недостаточна.
[membership level=»2,3″ show_noaccess=»true»]
Ввиду того, что зилебесиран селективно таргетирован против выработки ангиотензиногена в печени, оказываемые им альтерирующие эффекты на почечный ангиотензин II минимальны. Другими словами, почечные компенсаторные механизмы не затрагиваются. Это улучшает терапевтический индекс, позволяя назначать зилебесиран в повышенных дозах, не опасаясь развития неблагоприятных событий типа гипотензии, гиперкалиемии или острой почечной недостаточности, с которыми зачастую сталкиваются пациенты, принимающие стандартные гипотензивные препараты, блокирующие ренин-ангиотензин-альдостероновую систему (РААС), поскольку такие лекарства нацелены на нижележащие относительно ангиотензиногена компоненты сигнального пути РААС.
Опять же, зилебесиран позволит избавиться от избыточности в количестве используемых пациентами гипотензивных препаратов разных классов.
Следует, разумеется, тщательно изучить ряд важных вопросов, таких как не исключаемая потеря эффективности ввиду возможного появления антител к зилебесирану, его долгосрочная безопасность, более широкие преимущества в контексте сопутствующих заболеваний вроде сахарного диабета 2-го типа и хронической болезни почек.
Особняком стоит момент с возможностью быстрого отключения зилебесирана, если вдруг возникнет такая необходимость, к примеру, ввиду неожиданно сильного и устойчивого снижения артериального давления. Впрочем, создание малой интерферирующей РНК (миРНК), комплементарной миРНК зилебесирана особых трудностей не представляет.
Помимо грядущей опорной клинической программы фазы III, которая окончательно установит безопасность и эффективность зилебесирана среди широкой популяции пациентов, «Алнайлам фармасьютикалс» (Alnylam Pharmaceuticals) задумала проверить его способность снижать риск серьезных сердечно-сосудистых событий (MACE).
Предположительно, «Алнайлам» в начале коммерческого пути зилебесирана будет ориентировать его на лечение неконтролируемой гипертонии (она характеризуется скачкообразностью давления, недостаточным его снижением в ночное время, плохой приверженности пациентов терапии), которая устойчива к назначению трех или четырех гипотензивных препаратов разных классов. В дальнейшем, не исключено, будут подключены гипертоники с сердечной недостаточностью или диабетической нефропатией. В целом зилебесиран вполне способен стать новым стандартом лечения первичной (эссенциальной) гипертонии.
Коммерческий запуск зилебесирана на ключевых фармацевтических рынках планеты (включая США, Европу, Японию) ожидается приблизительно в 2030 году, причем с расширенным спектром терапевтических показаний, включающим в том числе снижение сердечно-сосудистой заболеваемости и смертности.
[/membership]
ЗИЛЕБЕСИРАН: КОЕ ЧТО ЕЩЕ
В свое время «Алнайлам фармасьютикалс» (Alnylam Pharmaceuticals) планировала разработку РНК-интерференционного препарата, в одной молекуле сочетающего две малые интерферирующие РНК (миРНК), чтобы одновременно осуществлять сайленсинг двух генов: ангиотензиногена (ANG) и ангиопоэтин-подобного белка 3 (ANGPTL3). Последний является валидированной мишенью в задаче снижения уровня атерогенных липидов.
Предполагаемый препарат, предназначенный для снижения риска серьезных сердечно-сосудистых событий (MACE) у высокорисковых пациентов, должен был снижать систолическое артериальное давление как минимум на 10 мм рт. ст. и уровень холестерина липопротеинов низкой плотности (ЛПНП) и триглицеридов хотя бы на 40%.
Параллельный сайленсинг реализовывался на рельсах технологической платформы GEMINI, а чрезвычайно редкое подкожное дозирование (один раз в год) — благодаря технологической платформе IKARIA.
Идея, высказанная еще в 2021 году, предполагала выбор препарата-кандидата в 2023 году. Однако, приоритеты, похоже, поменялись: ввиду бешеного спроса на препараты для лечения ожирения «Алнайлам» задумала заняться именно ими.
IONIS PHARMACEUTICALS И ПЕРЕДОВОЕ ЛЕЧЕНИЕ ГИПЕРТОНИИ
Вначале «Айонис фармасьютикалс» (Ionis Pharmaceuticals) пробовала разобраться с экспериментальным препаратом эвазарсен (evazarsen), который, по аналогии с зилебесираном (zilebesiran) авторства «Алнайлам фармасьютикалс» (Alnylam Pharmaceuticals), таргетирован против ангиотензиногена (AGT) в печени и предназначен для лечения резистентной гипертонии, не поддающейся адекватному контролю стандартными гипотензивными препаратами.
Затем, когда были собраны убедительные доказательства терапевтической состоятельности эвазарсена, «Айонис» переключилась на разработку его усовершенствованной версии в лице тонламарсена (tonlamarsen).
ЭВАЗАРСЕН: МЕХАНИЗМ ДЕЙСТВИЯ
Подкожно назначаемый эвазарсен (evazarsen, IONIS-AGT-LRx) представляет собой антисмысловой олигонуклеотид (ASO), ингибирующий синтез ангиотензиногена (AGT) в печени.
[membership level=»2,3″ show_noaccess=»true»]
Эвазарсен, будучи в точности комплементарным матричной РНК (мРНК), кодирующей AGT, связывается с ее нетранслируемым участком, тем самым приводя к ее же разрушению рибонуклеазой. В итоге предотвращается трансляция и останавливается синтез ангиотензиногена.
Для таргетной доставки эвазарсена в печень его ASO-последовательность ковалентно прилинкована к N-ацетилгалактозаминовому (GalNAc) лиганду, связывающему асиалогликопротеиновый рецептор 1 (ASGR1) на гепатоцитах.
В доклинических исследованиях эвазарсена на моделях грызунов с гипертонией или острой почечной недостаточностью было продемонстрировано падение уровня циркулирующего AGT на 90% с устойчивым снижением артериального давления [1].
Предполагалось, что коммерческая версия эвазарсена будет применяться подкожными инъекциями один или два раза в месяц.
[/membership]
ТОНЛАМАРСЕН: МЕХАНИЗМ ДЕЙСТВИЯ
«Айонис фармасьютикалс» (Ionis Pharmaceuticals), внимательно отслеживающая успехи конкурентов, поняла, что следует в срочном порядке оптимизировать режим дозирования эвазарсена (evazarsen), ведь прямой соперник зилебесиран (zilebesiran), которым занимается «Алнайлам фармасьютикалс» (Alnylam Pharmaceuticals), продемонстрировал, что эффективно снижает артериальное давление даже при очень редком дозировании подкожными инъекциями один раз в полгода.
[membership level=»2,3″ show_noaccess=»true»]
Для этого была подготовлена улучшенная версия эвазарсена — тонламарсен (tonlamarsen, ION904, AGT-2.5-LRx).
Антисмысловой олигонуклеотид (ASO) тонламарсен, во-первых, ингибирует синтез ангиотензиногена (AGT) в печени в более чем 10 раз сильнее, чем эвазарсен, и, во-вторых, может назначаться существенно реже: например, один раз в два или три месяца.
Не исключено, удастся разработать пероральную рецептуру тонламарсена.
[/membership]
ЭВАЗАРСЕН: КЛИНИЧЕСКИЕ РЕЗУЛЬТАТЫ
«Айонис фармасьютикалс» (Ionis Pharmaceuticals) завершила два клинических испытания NCT03714776 и NCT04083222 фазы II (рандомизированных, двойных слепых, плацебо-контролируемых, многоцентровых), которые оценили эффективность и безопасность эвазарсена (evazarsen) в лечении первичной (эссенциальной) артериальной гипертензии. Эвазарсен применялся соответственно монотерапевтически и в качестве дополнительного препарата к уже назначаемым гипотензивным лекарствам.
[membership level=»2,3″ show_noaccess=»true»]
Первое исследование охватило взрослых пациентов (n=25) с гипертонией (систолическое артериальное давление [САД] 140–165 мм рт. ст.), придерживающихся стабильной терапии двумя гипотензивными препаратами, один из которых — ингибитор ангиотензинпревращающего фермента (иАПФ) или блокатор рецептора ангиотензина II (БРА), а второй — бета-блокатор, блокатор кальциевых каналов или диуретик. Участники должны были прекратить прием всех гипотензивных лекарств.
Второе исследование пригласило пригласило взрослых пациентов (n=26) с неконтролируемой гипертонией (САД 140–170 мм рт. ст.), следующих стабильным курсом гипотензивной терапии из двух–трех препаратов разных классов, как то: в обязательном порядке иАПФ или БРА, плюс один–два дополнительных лекарства, таких как бета-блокатор, блокатор кальциевых каналов или диуретик (не относящийся к калийсберегающим). Участники должны были продолжать прием гипотензивных лекарств.
На протяжении 8 недель пациентам назначали 80-мг дозу эвазарсена или плацебо — подкожной инъекцией один раз в неделю.
Уровень сывороточного ангиотензиногена (AGT) в группах эвазарсена снизился на 54±25% и 67±14% в первом и втором исследованиях соответственно — против его роста на 3±18% и 13±23% в группах плацебо (p<0,001). Статистически значимое расхождение начало отмечаться на 8-й день, оставаясь таким вплоть до 92-го и 78-го дня.
Назначение эвазарсена привело к численно большему относительно плацебо (но статистически незначимому) снижению САД и диастолического артериального давления (ДАД). Так, усредненное изменение САД (мм рт. ст.) составило −8 (95% ДИ [здесь и далее]: −17, +2) и −12 (−21, −4), тогда как ДАД — −1 (−8, +5) и −6 (−11, −1).
В группах эвазарсена большая пропорция пациентов, чем в контрольных группах, продемонстрировала снижение САД и ДАД не менее чем на 5, 10 или 15 мм рт. ст., а также вышла к целевым показателям ниже 140 и 90 мм рт. ст. соответственно.
Второе исследование также показало отсутствие существенных различий между исходами в зависимости от того, принимали ли пациенты два (65% испытуемых) или три (35%) гипотензивных препарата.
Применение эвазарсена характеризовалось приемлемой переносимостью. Не зафиксировано гипотензивных событий, гиперкалиемии или почечных нарушений.
«Айонис» также запустила пару клинических испытаний фазы IIb (рандомизированных, двойных слепых, плацебо-контролируемых, многоцентровых), которые проверили еженедельное подкожное назначение эвазарсена в различных дозах и среди различных популяций пациентов.
Так, ASTRAAS (NCT04714320) изучило эвазарсен при неконтролируемой резистентной гипертонии среди пациентов (n=160), уже принимающих три и более гипотензивных препарата, а ASTRAAS-HF (NCT04836182) протестировало эвазарсен в лечении пациентов (n=72) с хронической сердечной недостаточностью со сниженной фракцией выброса (HFrEF).
Исследования завершены, однако их результаты не опубликованы.
[/membership]
ТОНЛАМАРСЕН: КЛИНИЧЕСКИЕ РЕЗУЛЬТАТЫ
«Айонис фармасьютикалс» (Ionis Pharmaceuticals) провела тонламарсен (tonlamarsen) через клиническое исследование NCT05314439 фазы II (рандомизированное, двойное слепое, плацебо-контролируемое, многоцентровое) среди взрослых пациентов (n=48) с неконтролируемой первичной (эссенциальной) артериальной гипертензией (систолическое артериальное давление [САД]) 130–170 мм рт. ст.), стабильно принимающих хотя бы одно гипотензивное лекарственное средство.
[membership level=»2,3″ show_noaccess=»true»]
Участникам назначали тонламарсен (30, 60 или 90 мг) или плацебо — подкожными инъекциями каждые 4 недели на протяжении 3 месяцев.
На 15-й неделе снижение уровня сывороточного ангиотензиногена (AGT) составило усредненных 79% и медианных 86% в 90-мг подгруппе тонламарсена — против его снижения на 6% и 5% в группе плацебо (p=0,001), тем самым механистически подтвердив состоятельность дозирования тонламарсена один раз в месяц [1].
Снижение САД как минимум на 10 мм рт. ст. было отмечено среди 42% пациентов, получавших тонламарсен, — против 30% в группе контроля.
Применение тонламарсена характеризовалось приемлемой переносимостью. Не зафиксировано ни целевых нежелательных явлений (НЯ), таких как гиперкалиемия, почечная дисфункция, гипотензия, ни внецелевых НЯ, таких как печеночная дисфункция, тромбоцитопения.
[/membership]
КОНВЕЙЕР
Сейчас конкуренции «Алнайлам фармасьютикалс» (Alnylam Pharmaceuticals) и ее зилебесирану (zilebesiran), считай, нет.
[membership level=»2,3″ show_noaccess=»true»]
С учетом того, насколько скромна «Айонис фармасьютикалс» (Ionis Pharmaceuticals) в объявлении клинических успехов, всё идет к тому, что программы эвазарсена (evazarsen) и тонламарсена (tonlamarsen) будут по итогам свернуты. Проблема состоит в том, что эти препараты-кандидаты, должным образом блокируя ангиотензиноген (AGT) в печени, требуют существенно более частого дозирования, чем зилебесиран, то есть не обладают конкурентным преимуществом.
Экспериментальный конвейер представлен считанными молекулами, таргетированными на AGT и изучаемыми в лечении гипертонии.
Так, раннестадийную клиническую проверку проходит РНК-интерференционный (RNAi) препарат-кандидат BW-00163 китайской «Арго байофарма» (Argo Biopharma). В начале 2024 года «Новартис» (Novartis) заключила с «Арго» многомиллиардное соглашение: не исключено, договоренности включают BW-00163 [1] [2].
Китайская «Сучжоу Райбо лайф сайенсиз» (Suzhou Ribo Life Science) занимается доклиническим RNAi-препаратом RBD9079 [3].
Китайские «Сучжоу Синеджин байо» (Suzhou Senegene Bio) и «Инновент байолоджикс» (Innovent Biologics) пробуют силы с доклиническим RNAi-препаратом SGB-3908 (IBI-3016) [4] [5].
На доклиническом этапе находятся STP136G и STP237G — RNAi-молекулы авторства «Сирнаомикс» (Sirnaomics) с офисами в США и Китае. Первое лекарственное соединение нацелено на AGT, второе — одновременно на AGT и аполипопротеин C3 (APOC3) [6].
«Вегови» (Wegovy, семаглутид) отныне можно применять для снижения риска серьезных нежелательных сердечно-сосудистых событий (сердечно-сосудистой смерти, нелетального инфаркта миокарда, нелетального инсульта) у взрослых пациентов с имеющимся сердечно-сосудистым заболеванием при наличии ожирения или избыточной массы тела.
Расширение списка показаний «Вегови», за которым стоит «Ново Нордиск» (Novo Nordisk), одобрено Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) в начале марта 2024 года.
«Вегови», появившийся в начале июня 2021 года, назначается еженедельными подкожными инъекциями для долгосрочной коррекции веса у пациентов в возрасте 12 лет и старше, страдающих либо ожирением, либо избыточной массой тела с сопутствующим лишнему весу заболеванием.
Семаглутид поможет сбросить 15% лишнего веса за год. И даже больше.
Ранее «Ново Нордиск» доказала, что «Вегови» успешно справляется с лечением сердечной недостаточности с сохраненной фракцией выброса среди пациентов с ожирением.
Семаглутид (semaglutide), агонист рецептора глюкагоноподобного пептида-1 (GLP1RA), влияет на широкий спектр метаболических путей, связанных с метаболизмом глюкозы, энергетическим гомеостазом и воспалением.
Дебют семаглутида состоялся в начале декабря 2017 года в лице препарата «Оземпик» (Ozempic, семаглутид), предназначенного для улучшения гликемического контроля при сахарном диабете 2-го типа. Впоследствии «Оземпик» продемонстрировал, что попутно снижает риск серьезных нежелательных сердечно-сосудистых событий при наличии сердечно-сосудистого заболевания и сдерживает прогрессирование существующей хронической почечной недостаточности.
Novo Nordisk предложила инъекционный семаглутид — агонист GLP-1, который эффективнее, чем «Трулисити».
Клинические подробности
Клиническое исследование SELECT (NCT03574597) фазы III (рандомизированное, двойное слепое, плацебо-контролируемое, многоцентровое, международное) проверило «Вегови» (Wegovy, семаглутид) или плацебо среди пациентов (n=17604) в возрасте 45 лет и старше с лишним весом или ожирением.
В анамнезе участников должно было быть сердечно-сосудистое заболевание, что подтверждалось хотя бы одним из следующих состояний:
перенесенный инфаркт миокарда;
перенесенный ишемический или геморрагический инсульт;
наличие симптоматической болезни периферических артерий, о которой свидетельствовала перемежающаяся хромота с лодыжечно-плечевым индексом в покое менее 0,85, пройденная процедура реваскуляризации периферических артерий или ампутация по причине атеросклеротического заболевания.
Композитная первичная конечная точка эффективности лечения была установлена временем до первого столкновения с серьезным сердечно-сосудистым событием (MACE), таким как сердечно-сосудистая смерть, нелетальный инфаркт миокарда или нелетальный инсульт.
После наблюдений в течение усредненных (39,8 ± 9,4) месяца, то есть в период приблизительно от 2,5 лет до 4 лет, события MACE были зарегистрированы для 6,5% и 8,0% пациентов в группах семаглутида (semaglutide) и плацебо.
Применение «Вегови» снизило риск MACE на относительных 20%: отношение риска (hazard ratio, HR) 0,80 (95% [здесь и далее]: 0,72–0,90; p<0,001).
Если говорить об отдельных компонентах MACE, назначение семаглутида привело к следующим снижениям рисков относительно плацебо:
сердечно-сосудистая смерть: на 15% (HR 0,85 [0,71–1,01]);
нелетальный инфаркт миокарда: на 28% (HR 0,72 [0,61–0,85]);
нелетальный инсульт: на 7% (HR 0,93 [0,74–1,15]).
Желудочно-кишечные расстройства, такие как тошнота, рвота и диарея, — наиболее распространенные нежелательные явления, которые привели к прекращению лечения семаглутидом.
Экспертные комментарии
Согласно прогнозам, к 2035 году у более половины населения планеты будет избыточная масса тела или ожирения [1]. В 2015 году высокий индекс массы тела (ИМТ) стал причиной 4 млн смертей, свыше двух третей из которых были вызваны сердечно-сосудистыми заболеваниями [2]. Лишний вес и ожирение ассоциированы с повышенным риском серьезных сердечно-сосудистых событий (MACE), причем даже после учета влияния метаболических факторов сердечно-сосудистого риска ввиду избыточного веса [3] [4] [5] [6].
Хотя снижение риска сердечно-сосудистых заболеваний путем лечения дислипидемии [7], гипертонии [8] и сахарного диабета [9] [10] является стандартной доказательной практикой, концепция лечения ожирения с целью снижения риска MACE сдерживается отсутствием должного набора клинических данных, указывающих на то, что модификация образа жизни или фармакологические вмешательства при избыточной массе тела или ожирении улучшают сердечно-сосудистые исходы [11] [12] [13] [14] [15].
Механизмы снижения сердечно-сосудистого риска при применении семаглутида (semaglutide) объясняются физиологическими преимуществами, которые получает организм после уменьшения количества избыточного жира
Во-первых, снижение веса приводит к улучшению уровня глюкозы и ослаблению традиционных факторов промежуточного риска сердечно-сосудистых заболеваний [16].
Во-вторых, уменьшение эктопических отложений жировой ткани благотворно сказывается на сдерживании прогрессирования атеросклероза и дисфункции миокарда [17], притом что периваскулярная и эпикардиальная жировая ткань оказывает прямое неблагоприятное воздействие на сосудистый эндотелий и миокард [18].
В-третьих, избавление от лишнего жира в организме улучшает системную провоспалительную и протромботическую картину, ассоциированную с ожирением [19].
Коррекция избыточной массы тела путем интенсивного изменения образа жизни (путем снижения калорийности рациона и усиленной физической активности) в целом не приводит к улучшению сердечно-сосудистых исходов [11] [14]. Связано это, возможно, с тем, что необходимо добиться снижения веса как минимум на 10% [20], а подобное весьма трудно достижимо без фармакологической поддержки. Напротив, бариатрическая хирургия, предполагающая похудение не менее чем на 20%, обеспечивает существенное снижение частоты MACE [21] [22].
Назначение семаглутида, изученное в этом клиническом испытании, помогло снизить вес в среднем на 9,4%, то есть не перешагнуло необходимый 10-процентный порог. Тем не менее семаглутид смог улучшить сердечно-сосудистые исходы. Есть мнение, что механизмы семаглутида, защищающие сердечно-сосудистую систему, привлекают множество взаимосвязанных путей, в том числе оказывающих последовательное влияние на кардиометаболические факторы риска.
Так, препараты класса агонистов рецептора глюкагоноподобного пептида-1 (GLP1RA), к которым относится семаглутид, в исследованиях на животных с сахарным диабетом и без него улучшили функции эндотелия и левого желудочка, способствовали стабильности атеросклеротических бляшек, снизили агрегацию тромбоцитов [23].
В этом клиническом испытании назначение семаглутида отразилось положительным изменением множества хорошо изученных биомаркеров сердечно-сосудистого риска, таких как артериальное давление, окружность талии, гликемический контроль, нефропатия, уровни липидов и C-реактивного белка. Что примечательно, указанные изменения были достигнуты на фоне применения статиновой терапии, гипотензивных лекарственных средств и прочих препаратов, используемых в лечении атеросклеротического сердечно-сосудистого заболевания.
Следует понимать, что в испытание были включены только пациенты с сердечно-сосудистыми заболеваниями, то есть остается неизвестным влияние семаглутида на первичную профилактику сердечно-сосудистых событий у лиц с избыточной массой тела или ожирением, но без атеросклеротического сердечно-сосудистого заболевания.
Ранее семаглутид подтвердил свою способность снижать риск MACE среди пациентов с сахарным диабетом 2-го типа при наличии сердечно-сосудистого заболевания или сердечно-сосудистых факторов риска: на 26% и 21% — соответственно при назначении семаглутида подкожными инъекциями еженедельно и перорально ежедневно [24] [25]. В этой популяции пациентов, согласно метаанализу, препараты GLP1RA-класса снижают риск MACE на 14% [10].