ЧТО ПРОИЗОШЛО
«Керендия» / «Фириалта» (Kerendia / Firialta, финеренон) — новый лекарственный препарат, предназначенный для лечения диабетической болезни почек.
ОСНОВНЫЕ ФАКТЫ
Пероральный финеренон (finerenone), разработанный «Байер» (Bayer), представляет собой первый в своем классе нестероидный антагонист минералокортикоидного рецептора.
«Керендия» одобрен Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) в начале июля 2021 года. Финеренон, назначаемый взрослым пациентам с хронической болезнью почек (ХБП), ассоциированной с сахарным диабетом 2-го типа (СД2), показан для снижения рисков стойкого снижения расчетной скорости клубочковой фильтрации, развития терминальной стадии болезни почек, сердечно-сосудистой смерти, нелетального инфаркта миокарда, госпитализации по поводу сердечной недостаточности [1].
В середине февраля 2022 года Европейское агентство по лекарственным средствам (EMA) выдало маркетинговое разрешение «Керендия» для лечения взрослых пациентов с ХБП (на стадии 3 и 4 с альбуминурией), ассоциированной с СД2. Через год европейский регулятор расширил инструкцию по применению финеренона, дозволив его применение в лечении ХБП (с альбуминурией), ассоциированной с СД2 [2].
В России финеренон получил регистрацию в конце марта 2023 года под брендом «Фириалта» (Firialta) для лечения ХБП (с альбуминурией) у взрослых с СД2 [3].

ПРЯМАЯ РЕЧЬ
«Пациенты, среди которых проводилось исследование финеренона, подвергались риску прогрессирования хронической болезни почек, несмотря на стандартное лечение, направленное на контроль над артериальным давлением и уровнем глюкозы в крови. Теперь у них есть новый метод лечения, обеспечивающий защиту почек».
Джордж Бакрис (George Bakris), ведущий автор исследования, основатель и директор комплексного центра гипертонии при Медицинском центре Чикагского университета (Чикаго, шт. Иллинойс, США).
«Пациенты с хронической болезнью почек сталкиваются с повышенным риском сердечно-сосудистых событий. И поскольку она прогрессирует незаметно и на ранних стадиях зачастую не сопровождается симптомами, пациенты с сахарным диабетом 2-го типа должны регулярно наблюдаться у врача в целях выявления первых признаков заболевания почек, после постановки диагноза которого им следует проходить комплексное лечение для снижения риска сердечно-сосудистых осложнений и смерти».
Петер Россинг (Peter Rossing), соавтор исследования и руководитель направления осложнений в Центре имени Стено (Копенгаген, Дания).
«Хроническая болезнь почек, ассоциированная с сахарным диабетом 2-го типа, оказывает весьма изнурительное влияние на жизнь пациентов. Эта болезнь имеет далеко идущие последствия, вот почему важно иметь новые варианты лечения, которые замедляют ее прогрессирование».
Кевин Лонгино (Kevin Longino), исполнительный директор Национального нефрологического фонда США (NKF, Нью-Йорк, США) — некоммерческой организации, занимающаяся информированием, профилактикой и лечением заболеваний почек.
«Финеренон — первый и единственный нестероидный антагонист минералокортикоидных рецепторов, доказавший свою способность значительно замедлять прогрессирование хронической болезни почек и снижать сердечно-сосудистый риск у людей с хронической болезнью почек, ассоциированной с сахарным диабетом 2-го типа».
Амит Шарма (Amit Sharma), вице-президент по сердечно-сосудистым и почечным медицинским вопросам «Байер» (Bayer).
«Даже когда уровень глюкозы в крови и артериальное давление хорошо контролируются, риск прогрессирования заболевания почек и сердечно-сосудистых событий остается высоким у пациентов с хронической болезнью почек и сахарным диабетом 2-го типа. Финеренон устраняет ключевую причину прогрессирования заболевания, на которую не воздействуют другие препараты. Финеренон предлагает особый способ защиты пациентов от дальнейшего повреждения почек и сердечно-сосудистых событий».
Майкл Девой (Michael Devoy), медицинский директор фармацевтического подразделения «Байер» (Bayer).
СУТЬ ВОПРОСА
СД2 является основной причиной ХБП во всём мире [1]. Насчитывается свыше 200 млн пациентов, которые одновременно страдают ХБП и диабетом [2] [3] [4], и в паре эти заболевания сокращают продолжительность жизни на 16 лет [5]. До 40% диабетиков в конечном итоге заболевают ХБП [6].
Международные рекомендации по лечению ХБП у пациентов с СД2 рекомендуют контролировать гипертонию и гипергликемию, а также использовать блокаторы ренин-ангиотензин-альдостероновой системы (РААС), такие как ингибиторы ангиотензинпревращающего фермента (иАПФ) и блокаторы ангиотензиновых рецепторов (БРА). Недавно добавились рекомендации по применению ингибиторов натрий-глюкозного котранспортера 2-го типа (SGLT2) [7] [8]. Тем не менее, несмотря на рекомендованное лечение, риск прогрессирования ХБП сохраняется [9], и потому необходимы новые методы лечения.

Семаглутид для лечения диабетической болезни почек
Семаглутид — новый стандарт лечения хронической болезни почек при сахарном диабете 2-го типа.
КАК ЭТО РАБОТАЕТ
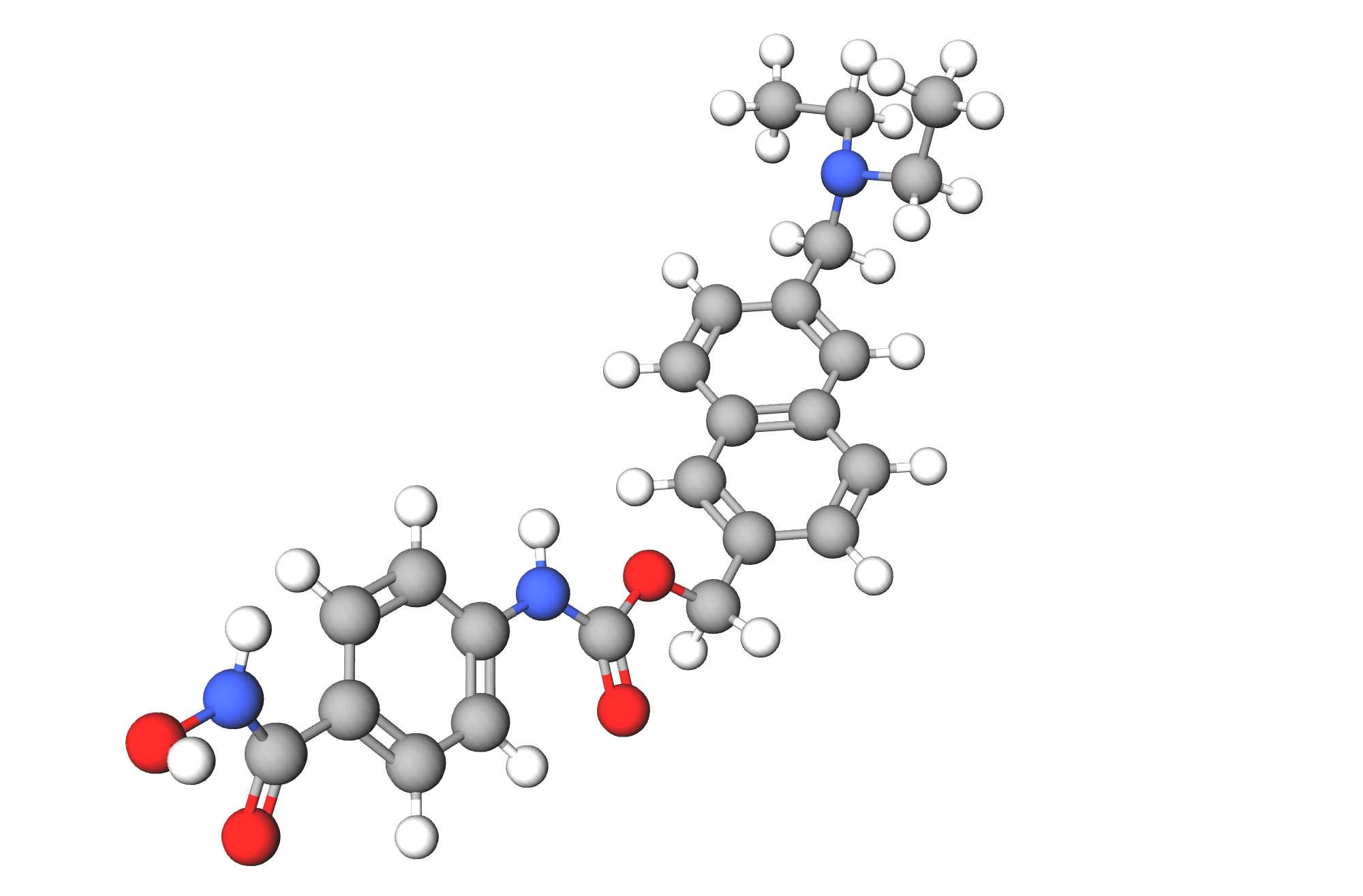
Финеренон (finerenone, BAY 94-8862) — пероральный низкомолекулярный высокоселективный нестероидный антагонист минералокортикоидного рецептора (MR).
Данные свидетельствуют о патофизиологической роли чрезмерной активации минералокортикоидных рецепторов при кардиоренальных заболеваниях, включая ХБП и диабет: опосредованные ею воспаление и фиброз приводят к прогрессирующей дисфункции почек и сердечно-сосудистой системы [1] [2] [3] [4].
Подавление сверхактивности MR, вызванной альдостероном и кортизолом, приводит к сдерживанию патологических процессов воспаления, фиброза, роста артериального давления, желудочковой гипертрофии.
Финеренон — в отличие от всех существующих MR-блокаторов, таких как спиронолактон (spironolactone), эплеренон (eplerenone) или канренон (canrenone) — характеризуется уникальным фармакологическим профилем [5].
Во-первых, финеренон никак не затрагивает глюкокортикоидные, андрогеновые, прогестероновые и эстрогеновые рецепторы, тем самым избавляя от расхожих при назначении MR-антагонистов нежелательных явлений вроде гинекомастии, импотенции, снижения либидо.
Во-вторых, финеренон не относится к стероидным средствам (он является производным дигидропиридина), что позволяет ему с высокой аффинностью связываться с MR и более эффективно подавлять транскрипционные коактиваторы, участвующие в экспрессии гипертрофических и профибротических генов.
В-третьих, финеренон относительно равномерно распределяется между сердечной и почечной тканями.
В доклинических моделях финеренон оказывал более мощное противовоспалительное и антифибротическое действие, чем стероидные MR-антагонисты [6] [7] [8] [9].
Финеренон снижал соотношение альбумина и креатинина в моче у пациентов с ХБП, получающих РААС-блокаторы, при этом он оказывал меньшее влияние на уровень калия в сыворотке крови, чем спиронолактон [10] [11].
КЛИНИЧЕСКИЕ ПОДРОБНОСТИ
«Байер» осуществила две масштабных клинических проверки финеренона при диабетической нефропатии. Одно клиническое испытание изучило препарат «Керендия» / «Фириалта» среди страдающих СД2 с запущенной ХБП (главным образом на стадии 3 или 4 и с тяжелой альбуминурией). Второе исследование протестировало «Керендия» / «Фириалта» среди диабетиков с ХБП в менее тяжелой форме (либо на стадии 2–4 и с умеренной альбуминурией, либо на стадии 1–2 и с тяжелой альбуминурией).
FIDELIO-DKD
Клиническое исследование FIDELIO-DKD (NCT02540993) фазы III (рандомизированное, двойное слепое, плацебо-контролируемое, многоцентровое, международное) пригласило взрослых пациентов (n=5674) с диабетической нефропатией, характеризующейся персистирующей высокой альбуминурией.
Среди основных критериев включения:
- сахарный диабет 2-го типа;
- хроническая болезнь почек;
- либо стойкая высокая альбуминурия (соотношение альбумина и креатинина в моче [UACR] в пределах 30–300 мг/г), расчетная скорость клубочковой фильтрации (eGFR) в диапазоне 25–60 мл/мин/1,73 м2 и диабетическая ретинопатия, либо стойкая очень высокая альбуминурия (UACR ≥ 300 мг/г) и eGFR в диапазоне 25–75 мл/мин/1,73 м2;
- предшествовавшее лечение ингибиторами ангиотензинпревращающего фермента (иАПФ) и/или блокаторами ангиотензиновых рецепторов (БРА);
- уровень калия в сыворотке ≤ 4,8 ммоль/л.
Среди основных характеристик испытуемых:
- средний возраст 66 лет, 70% мужчин;
- средняя eGFR 44 мл/мин/1,73 м2, у 55% eGFR < 45 мл/мин/1,73 м2;
- медиана UACR 852 мг/г (межквартильный размах [IQR] 446–1634);
- у 46% атеросклеротическое сердечно-сосудистое заболевание в анамнезе;
- 99,8% получали иАПФ или БРА;
- 97% проходили противодиабетическую терапию: 64% получали инсулин, 44% метформин, 7% агонисты рецептора глюкагоноподобного пептида-1 (GLP1RA), 5% ингибиторы натрий-глюкозного котранспортера 2-го типа (SGLT2);
- 74% принимали статины, 57% антиагреганты.
Участники ежедневно получали плацебо или финеренон поверх стандартной терапии, предполагавшей назначение гипогликемических лекарственных средств и максимально переносимой дозы иАПФ или БРА.
В ходе наблюдений медианных 2,6 года финеренон замедлил прогрессирование хронической болезни почек, обеспечив статистически значимую отсрочку времени до наступления первого случая почечной недостаточности, стабильного снижения eGFR (на 40% и более за период минимум 4 недель подряд) или смерти из-за отказа почек. Уменьшение вероятности указанного композитного риска составило 18% по сравнению с плацебо: отношение риска (hazard ratio, HR) 0,82 (95% ДИ [здесь и далее]: 0,73–0,93; p=0,001) [1].
Если говорить об уменьшении рисков отдельных событий, то для почечной недостаточности оно получилось равным 13% (HR 0,87 [0,72–1,05]), а для стабильного снижения eGFR — 19% (HR 0,81 [0,72–0,92]).
«Керендия» / «Фириалта» статистически значимым образом ослабил композитный риск сердечно-сосудистых событий: кардиоваскулярной смерти, нелетального инфаркта миокарда, нелетального инсульта или госпитализации по причине сердечной недостаточности: на 14% относительно плацебо (HR 0,86 [0,75–0,99; p=0,034]).
При этом частота отдельных событий была ниже при применении финеренона, чем плацебо, за исключением нелетального инсульта, частота которого была одинаковой в обеих группах. Так, отношение риска для сердечно-сосудистой смерти, нелетального инфаркта миокарда, нелетального инсульта и госпитализации по причине сердечной недостаточности вышло к следующим показателям: 0,86 (0,68–1,08), 0,80 (0,58–1,09), 1,03 (0,76–1,38) и 0,86 (0,68–1,08).
Среди наиболее распространенных нежелательных явлений (НЯ) в ответ на назначение препарата «Керендия» / «Фириалта»: гиперкалиемия (у 18,3% пациентов — против 9,0% в группе плацебо), гипотония (4,8% против 3,4%), гипонатриемия (1,4% против 0,7%). Гиперкалиемия обусловила полное прекращение лечения 2,3% испытуемых — против 0,9%. С госпитализацией по причине гиперкалиемии столкнулись 1,4% участников, получавших финеренон, — против 0,3% в контрольной группе.




FIGARO-DKD
Клиническое исследование FIGARO-DKD (NCT02545049) фазы III (рандомизированное, двойное слепое, плацебо-контролируемое, многоцентровое, международное) пригласило взрослых пациентов (n=7352) с диабетической нефропатией, характеризующейся персистирующей высокой альбуминурией.
Основные критерии включения идентичны таковым в FIDELIO-DKD, разве что было одно отличие:
- либо стойкая высокая альбуминурия (UACR в пределах 30–300 мг/г) и eGFR в диапазоне 25–90 мл/мин/1,73 м2, либо стойкая очень высокая альбуминурия (UACR ≥ 300 мг/г) и eGFR минимум 60 мл/мин/1,73 м2.
Среди основных характеристик испытуемых:
- средний возраст 64 года, 69% мужчин;
- средняя eGFR 68 мл/мин/1,73 м2, у 62% eGFR ≥ 60 мл/мин/1,73 м2, у 83% eGFR ≥ 45 мл/мин/1,73 м2;
- медиана UACR 308 мг/г (IQR 108–740);
- 99,9% получали иАПФ или БРА;
- 98% проходили противодиабетическую терапию: 54% получали инсулин, 8% агонисты GLP-1, 8% ингибиторы SGLT2;
- 71% принимали статины, 48% диуретики.
Участники ежедневно получали плацебо или финеренон поверх стандартной терапии, предполагавшей назначение гипогликемических лекарственных средств и максимально переносимой дозы иАПФ или БРА.
В ходе наблюдений медианных 3,4 года финеренон статистически значимым образом снизил риск выхода к композитной конечной точке, представленной одним из сердечно-сосудистых событий, таких как сердечно-сосудистая смерть, нелетальный инфаркт миокарда, нелетальный инсульт, госпитализация по причине сердечной недостаточности. Уменьшение вероятности указанного композитного риска составило 13% по сравнению с плацебо: HR 0,87 (0,76–0,98; p=0,03) [1].
Частота отдельных событий была ниже при применении финеренона, чем плацебо: отношение риска для сердечно-сосудистой смерти, нелетального инфаркта миокарда, нелетального инсульта и госпитализации по причине сердечной недостаточности вышло к следующим показателям: 0,90 (0,74–1,09), 0,99 (0,76–1,31), 0,97 (0,74–1,26) и 0,71 (0,56–0,90).
«Керендия» / «Фириалта» на 13% уменьшил вероятность композитного риска, составленного из таких событий, как почечная недостаточность, стабильное снижение eGFR (на 40% и более за период минимум 4 недель подряд) или смерть из-за отказа почек: HR 0,87 (0,76–1,01).
Снижение риска (HR) отдельных событий оказалось следующим: почечная недостаточность 0,72 (0,49–1,05), терминальная стадия болезни почек 0,64 (0,41–0,995), стабильное снижение eGFR до < 15 мл/мин/1,73 м2 0,71 (0,43–1,16), стабильное снижение eGFR на ≥ 40% 0,87 (0,75–1,00), стабильное снижение eGFR на ≥ 57% 0,76 (0,58–1,00).
Среди наиболее распространенных НЯ в ответ на назначение препарата «Керендия» / «Фириалта» была гиперкалиемия (у 10,8% пациентов — против 5,3% в группе плацебо). Уровень сывороточного калия выше 5,5 ммоль/л зарегистрирован у 13,5% против 6,4%, выше 6,0 ммоль/л — у 2,3% против 1,2%. Гиперкалиемия обусловила полное прекращение лечения 1,2% испытуемых против 0,4%. С госпитализацией по причине гиперкалиемии столкнулись 0,6% пациентов, получавших финеренон, — против 0,1% в контрольной группе.


ЧТО ДАЛЬШЕ
«Байер», намеревающаяся расширить популяцию пригодных пациентов, продолжает изучение препарата «Керендия» / «Фириалта» в следующих клинических испытаниях фазы III:
- FIND-CKD (NCT05047263) среди взрослых (n=1584) с ХБП, не ассоциированной с СД2: по силам ли финеренону замедлить прогрессирование недиабетической болезни почек;
- FINE-ONE (NCT05901831) среди взрослых (n=220) с сахарным диабетом 1-го типа и ХБП: насколько хорошо финеренон оказывает нефропротекторное действие;
- FIONA (NCT05196035) среди педиатрических (от 6 месяцев до 17 лет) пациентов (n=219) с ХБП и протеинурией: справится ли финеренон с замедлением прогрессирования ХБП;
- FIONA-OLE (NCT05457283) среди педиатрических (от 1 года до 18 лет) пациентов (n=100) с ХБП и протеинурией: долгосрочная оценка безопасности и эффективности финеренона, назначаемого на протяжении полутора лет.
Клиническое исследование CONFIDENCE (NCT05254002) фазы II рассматривает комбинацию финеренона с SGLT2-ингибитором «Джардинсом» (Jardiance, эмпаглифлозин) среди взрослых (n=807): превосходит ли комбинированное лечение СД2-ассоциированной ХБП ее монотерапию финереноном или эмпаглифлозином.
Отдельно «Байер» проверяет финеренон в лечении сердечной недостаточности, и уже достигла определенных успехов.

«Керендия» / «Фириалта»: финеренон против сердечной недостаточности
Финеренон — новое лечение сердечной недостаточности с умеренно сниженной или сохраненной фракцией выброса.
ЭКСПЕРТНЫЕ КОММЕНТАРИИ
Хроническая болезнь почек (ХБП) усугубляет сердечно-сосудистый риск, ассоциированный с сахарным диабетом 2-го типа (СД2) [1].
Риск сердечно-сосудистых событий и впервые возникшей сердечной недостаточности возрастает по мере того, как соотношение альбумина и креатинина в моче (UACR) превышает 10 мг/г, а расчетная скорость клубочковой фильтрации (eGFR) падает ниже 75 мл/мин/1,73 м2 [1] [2] [3] [4]. У большинства пациентов с ХБП риск развития сердечно-сосудистых событий выше, чем риск развития почечной недостаточности [5]. Вот почему важно выявлять и лечить ХБП, чтобы снизить высокое бремя сердечно-сосудистых заболеваний и сердечной недостаточности, связанное с ХБП у пациентов с СД2 [1] [6].
Тем не менее пациенты с высоким показателем UACR, но нормальной eGFR не всегда выявляются из-за того, что анализ UACR не проводится на регулярной основе, хотя и является золотым стандартом для идентифицирования повреждения почек. И это проблема как для больных, которых должным образом не лечат, так и для «Байер», которая заинтересована в продвижении финеренона.
Долгосрочные клинические испытания финеренона, запущенные в 2015 году, проводились в тот момент, когда ингибиторы натрий-глюкозного котранспортера 2-го типа (SGLT2) начали выходить за пределы лечения исключительно диабета. Так, в конце сентября 2019 года «Инвокана» (Invokana, канаглифлозин), за которым стоит «Янссен» (Janssen) в составе «Джонсон энд Джонсон» (Johnson & Johnson), был одобрен для снижения рисков развития терминальной стадии болезни почек, удвоения уровня сывороточного креатинина, сердечно-сосудистой смерти и госпитализации по поводу сердечной недостаточности у взрослых пациентов с СД2 и диабетической нефропатией с альбуминурией.
Затем, в конце апреля 2021 года, «Фарсига» / «Форсига» (Farxiga / Forxiga, дапаглифлозин), предлагаемый «АстраЗенека» (AstraZeneca), был разрешен для снижения рисков стойкого снижения eGFR, смерти от сердечно-сосудистых осложнений на терминальной стадии ХБП и госпитализации по поводу сердечной недостаточности у взрослых пациентов с ХБП и риском ее прогрессирования.
Указанные SGLT2-ингибиторы установили высокую планку терапевтической эффективности при ХБП, и перешагнуть ее финеренон не смог. Если сравнить соответствующие клинические исходы препаратов «Керендия» / «Фириалта», «Инвокана» и «Фарсига» / «Форсига», всё свидетельствует не в пользу финеренона. Следует, впрочем, понимать, что подобные сравнения зачастую методологически некорректны ввиду различий в популяциях пациентов и протоколов клинических испытаний.
Есть мнение, что благодаря различающимся механизмам действия финеренон уместно применять совместно с SGLT2-ингибиторами или GLP1RA-препаратами, к примеру «Оземпиком» (Ozempic, семаглутид) или «Трулисити» (Trulicity, дулаглутид), которые разработали «Ново Нордиск» (Novo Nordisk) и «Илай Лилли» (Eli Lilly) и которые несут явную сердечно-сосудистую пользу. Предполагалось, что «Байер» вряд ли согласится на клинические исследования подобных комбинаций: нужны огромные ресурсы, чтобы выявить, возможно, не столь значительный аддитивный эффект, притом что патентная защита финеренона истекает в 2029 году. Однако немецкий химико-фармацевтический гигант на это всё же пошел.

Семаглутид и тирзепатид: эффективное лечение сердечной недостаточности
«Оземпик», «Вегови», «Мунджаро», «Зепбаунд» — для здоровья сердца.
БИЗНЕС
Согласно отраслевым прогнозам, сделанным до появления финеренона на рынке, к 2026 году он должен выйти на уровень ежегодных продаж в 1,2 млрд долларов: благодаря равно как тому, что «Керендия» / «Фириалта» займет должное место в фармакологическом арсенале борьбы с диабетической болезнью почек, так и расширению списка своих показаний.
Фактический заработок финеренона в 2022 и 2023 гг. составил 107 млн евро ($112 млн) и €270 млн ($292 млн). В первой половине 2024-го «Керендия» / «Фириалта» принес €200 млн ($216 млн).