Экспериментальный нерандомиласт (nerandomilast) успешно справился с позднестадийной клинической проверкой лечения идиопатического легочного фиброза (ИЛФ).
ОСНОВНЫЕ ФАКТЫ
«Бёрингер Ингельхайм» (Boehringer Ingelheim) успешно провела пероральный препарат-кандидат нерандомиласт через опорное клиническое испытание фазы III.
Применение нерандомиласта обеспечило улучшение функции легких.
В ближайшее время в адрес регуляторов будет направлено досье для регистрации нерандомиласта.
ПРЯМАЯ РЕЧЬ
«Нерандомиласт сдерживает темп снижения функции легких, и делает это равно как у тех пациентов, кто уже принимает противофиброзные препараты, так и у тех, кто ими не пользуется».
Лука Ричелди (Luca Richeldi), глава пульмонологического отделения Университетского поликлинического фонда имени Агостино Джемелли (Fondazione Policlinico Universitario Agostino Gemelli) при Католическом университете Пресвятого Сердца (Università Cattolica del Sacro Cuore, UCSC, Рим, Италия), ведущий автор исследования.
«Это первое за последнее десятилетие клиническое испытание фазы III экспериментального лечения идиопатического легочного фиброза, которому удалось добраться до первичной конечной точки».
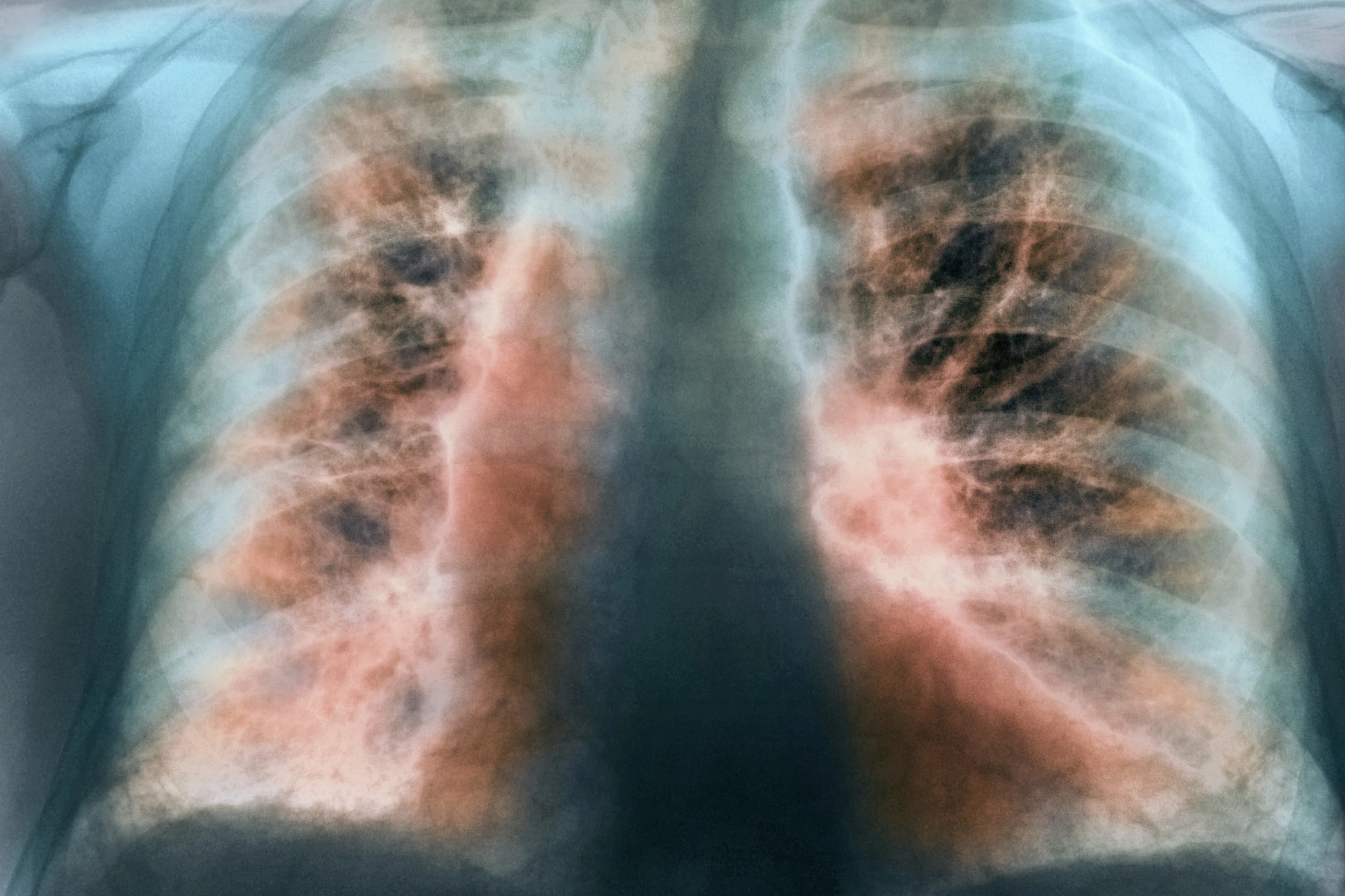
Идиопатический легочный фиброз (ИЛФ) — прогрессирующее и необратимое заболевание легких с высокой летальностью, сопровождающееся такими симптомами, как одышка во время физической активности, сухой и постоянный кашель, дискомфорт в груди, усталость и слабость [1] [2].
Нынешнее лечение ИЛФ осуществляется препаратами «Офев» / «Варгатеф» (Ofev / Vargatef, нинтеданиб) и «Эсбриет» (Esbriet, пирфенидон), которые замедляют, но не останавливают прогрессирование фиброза [3] [4] [5].
Нинтеданиб (nintedanib), продвигаемый «Бёрингер Ингельхайм» (Boehringer Ingelheim), представляет собой конкурентный ингибитор множества нерецепторных и рецепторных тирозинкиназ, тогда как противофиброзный механизм действия пирфенидона (pirfenidone), за которым стоит «Рош» (Roche), по-прежнему остается до конца невыясненным.
В любом случае лечение идиопатического легочного фиброза оставляет желать много лучшего [6].
БОЛЬШИЕ ЧИСЛА
Идиопатический легочный фиброз (ИЛФ) — одно из наиболее распространенных прогрессирующих фиброзирующих интерстициальных болезней легких (ИЛД). Хотя ИЛФ считается редким (орфанным) заболеванием, им страдают приблизительно 3 млн человек во всём мире. Заболевание в основном поражает пациентов старше 50 лет и чаще мужчин, чем женщин [1] [2].
Известно, что ингибирование фосфодиэстеразы 4 (PDE4) ассоциировано с противовоспалительными и противофиброзными эффектами: первые достигаются за счет уменьшения высвобождения противовоспалительных медиаторов и сдерживания рекрутинга воспалительных клеток, вторые — ослабления фиброзного ремоделирования [1] [2] [3] [4].
Однако селективное ингибирование PDE4B всё же оптимальнее, поскольку оно равно как наделено такими же терапевтически полезными свойствами [5] [6], так и несет за собой более приемлемый профиль безопасность, если сравнивать с неизбирательным ингибированием PDE4 [1] [6], которое сопровождается такими нежелательными явлениями (НЯ), как тошнота, рвота и диарея [7].
Указанные НЯ обусловлены, как предполагается, с ингибированием PDE4D [8]. Есть мнение, что избирательное ингибирование PDE4B их нивелирует: нерандомиласт приблизительно в 10 раз более селективен в отношении ингибирования PDE4B, чем PDE4D [7].
КЛИНИЧЕСКИЕ ПОДРОБНОСТИ
FIBRONEER-IPF
Клиническое испытание FIBRONEER-IPF (NCT05321069) фазы III (рандомизированное, двойное слепое, плацебо-контролируемое, многоцентровое, международное) проверило назначение нерандомиласта (9 или 18 мг) или плацебо (два раза в день) взрослым (40 лет и старше) пациентам (n=1177) с идиопатическим легочным фиброзом. Участникам было дозволено продолжать придерживаться фоновой терапии нинтеданибом или пирфенидоном, если таковая проводилась [1].
По прошествии 52 недель лечения группа нерандомиласта продемонстрировала статистически и клинически значимое превосходство над группой плацебо в том, что касается абсолютного увеличения объема форсированного выдоха за 1-ю секунду (ОФВ1) [2].
О результатах, относящихся к другим конечным точкам эффективности лечения, таким как время до первого обострения заболевания, время до первой госпитализации по респираторным причинам, время до смертельного исхода, ничего не сказано.
Все подробности будут раскрыты в первой половине 2025 года.
NCT04419506
В предшествовавшем 12-недельном клиническом исследовании NCT04419506 фазы II среди взрослых пациентов (n=147) с идиопатическим легочным фиброзом применение нерандомиласта дважды в день обеспечило изменение ОФВ1 на медианных +5,7 мл (95% ДИ [здесь и далее]: −39,1, +50,5) и +2,7 мл (−32,8, +38,2) в подгруппах пациентов, соответственно не получавших фоновую противофиброзную терапию и получавших таковую, — против изменения на −81,7 мл (−133,5, −44,8) и −59,2 мл (−111,8, −17,9) в подгруппах плацебо [1] [2].
Среди наиболее распространенных нежелательных явлений в ответ на использование нерандомиласта: диарея, проявившаяся по большей части в легкой форме. С ней столкнулись до трети участников.
ЧТО ДАЛЬШЕ
«Бёрингер Ингельхайм» собирается отправить в адрес регуляторов регистрационное досье нерандомиласта.
ЧТО ЕЩЕ
«Бёрингер Ингельхайм» параллельно осуществляет клиническую проверку FIBRONEER-ILD (NCT05321082) фазы III нерандомиласта в лечении прогрессирующего легочного фиброза при иных интерстициальных болезнях легких [1].
Таких паренхиматозных легочных заболеваний насчитывается свыше 200, причем большинство из них относятся к редким, как то: идиопатическая неспецифическая интерстициальная пневмония, респираторный бронхит, ассоциированный с интерстициальным заболеванием легких, десквамативная интерстициальная пневмония, криптогенная организующаяся пневмония, острая интерстициальная пневмония, неклассифицируемая идиопатическая интерстициальная пневмония, саркоидоз, гиперчувствительный пневмонит, лимфангиолейомиоматоз, лангергансоклеточный гистиоцитоз и др.
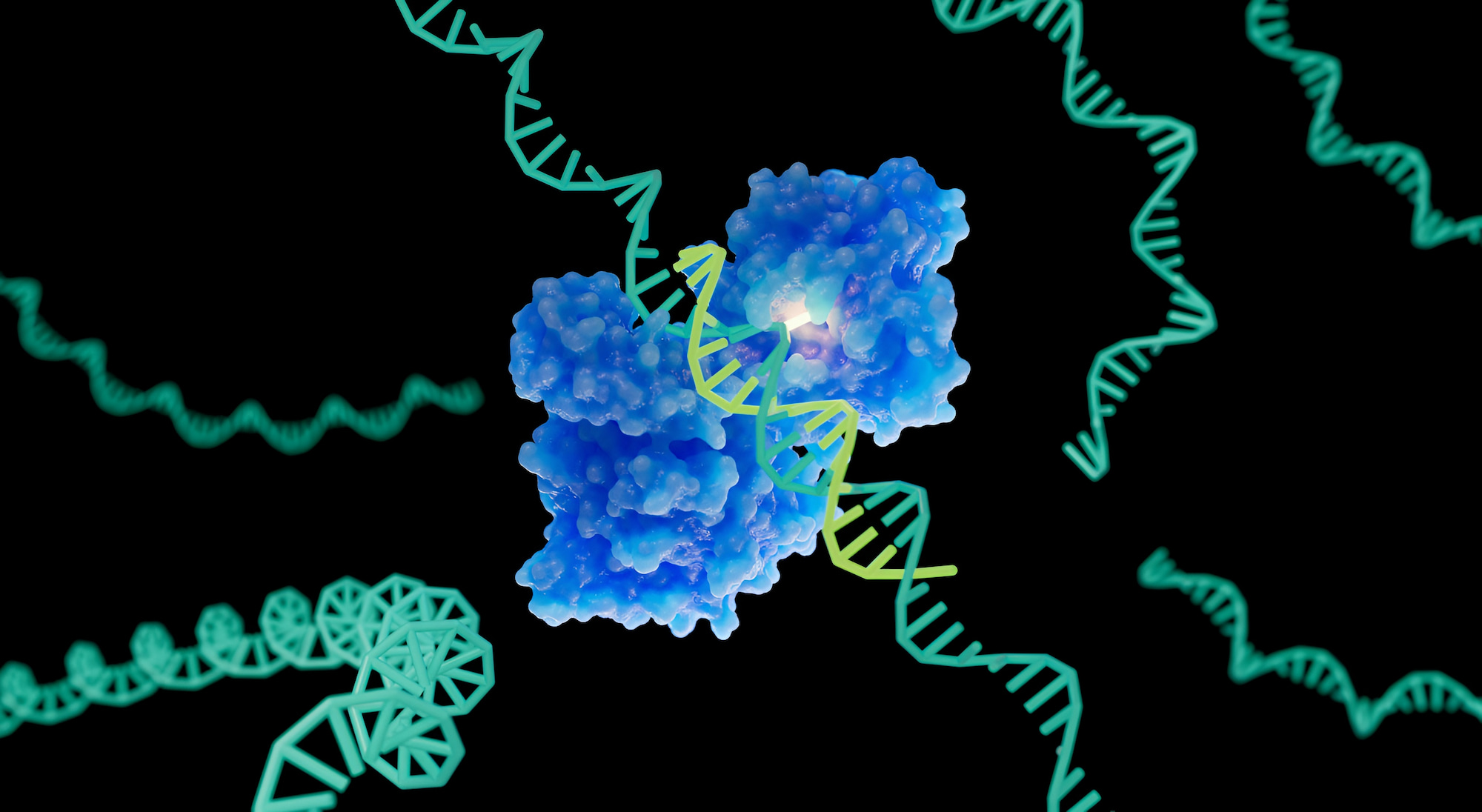
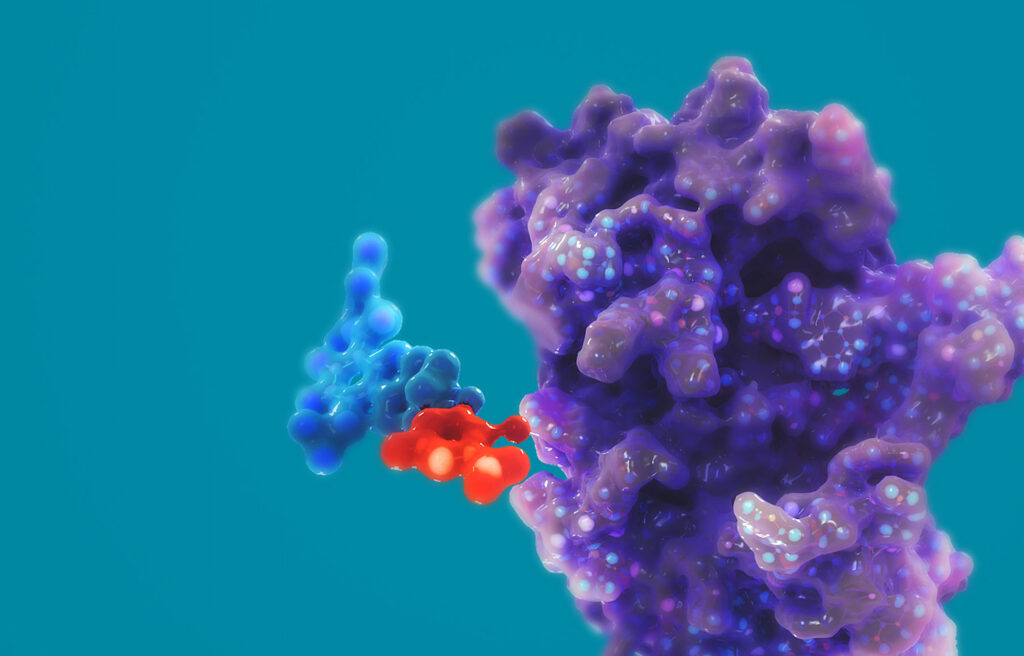
Впервые в истории медицины и биотехнологий удалось добиться успешного терапевтического редактирования РНК непосредственно в организме человека.
ОСНОВНЫЕ ФАКТЫ
«Вейв лайф сайенсиз» (Wave Life Sciences) доказала состоятельность механизма действия экспериментального олигонуклеотида, у которого получилось исправить генетический дефект, ответственный за недостаточность альфа-1-антитрипсина.
Подкожно вводимый препарат-кандидат WVE-006 меняет, считай, «одну букву» в наборе инструкций мРНК, которые должны кодировать синтез функционального белка, но который из-за мутации не вырабатывается должным образом.
Терапевтическая модальность, предложенная «Вейв», в корне отличается от подходов, обращающихся к генной терапии и генному редактированию, и характеризуется рядом преимуществ перед ними.
Так, редактирование мРНК не сопряжено с риском неправильного изменения генов человека, которое может стать необратимым: поскольку измененные мРНК быстро разрушаются в организме, результаты редактирования носят временный характер, что облегчает прекращение терапии и уменьшение побочных эффектов. Поскольку при таком подходе не используются бактериальные ферменты, такие как Cas9 в ходе генного редактирования CRISPR, отсутствуют риски провоцирования иммунной системы, которая должна ответить на чужеродные белки. Доставка мРНК-редактирующих препаратов в организм не требует сторонних систем вроде вирусных векторов или липидных наночастиц.
Впрочем, не всё так радужно. Во-первых, по-прежнему нельзя целиком и полностью исключать внецелевых эффектов. Во-вторых, подход «Вейв» не является универсально подходящим для решения любых генетических поломок.
На волне положительный известий биржевые котировки «Вейв» прибавили 75%.
Коммерческие права на WVE-006 принадлежат «ГлаксоСмитКляйн» (GlaxoSmithKline), заблаговременно оформившей партнерство с «Вейв».
ПРЯМАЯ РЕЧЬ
«Первое в истории успешное терапевтическое редактирование РНК человека — важная веха для всей области лекарств на базе олигонуклеотидов. Это достижение подтверждает мощный потенциал нашей фирменной биотехнологической платформы, которой под силу предложить качественно новые препараты для лечения самых разнообразных заболеваний, в том числе недостаточности альфа-1-антитрипсина, мышечной дистрофии Дюшенна, болезни Гентингтона, ожирения».
Пол Болно (Paul Bolno), президент и исполнительный директор «Вейв лайф сайенсиз» (Wave Life Sciences).
Патология характеризуется дефицитом функционально полноценных белков AAT, являющихся протеазными ингибиторами, и отражается гибелью гепатоцитов, воспалением печени, нарушением ее регенерации и прогрессирующим фиброзом, который ведет к циррозу. Помимо болезни печени заболевание манифестирует панацинозной эмфиземой легких, панникулитом, васкулитом.
И если недостаточность альфа-1-антитрипсина, вызывающая болезнь легких, может лечиться внутривенной заместительной терапией путем еженедельного вливания экзогенного AAT, полученного из плазмы доноров, то лекарств против ассоциированной болезни печени пока не предложено. Разве что можно провести процедуру трансплантации печени.
Идентифицировано свыше 150 мутаций, затрагивающих ген SERPINA1. Самой распространенной причиной тяжелого дефицита AAT является точечная мутация Glu342Lys, представляющая собой замену одной пары оснований, гуанина на аденин (G-to-A), что приводит к замене глутаминовой кислоты на лизин в положении 342 [5].
Нормальный аллель гена SERPINA1 обозначается буквой M. Поскольку ААТ является ингибитором протеазы, обозначение Pi означает «ингибитор протеазы», а буквы — присутствующие аллели. Так, Pi*MM означает гомозиготность по нормальному гену, а Pi*ZZ — гомозиготность по аллелю Z; под последним понимают мутацию Glu342Lys.
БОЛЬШИЕ ЧИСЛА
Хотя недостаточность альфа-1-антитрипсина обычно считается редким заболеванием, однако, судя по оценкам, более 3 млн человек во всём мире являются носителями комбинации аллелей, которая способна привести к тяжелому дефициту ААТ [1] [2].
Считается, что диагноз недостаточности альфа-1-антитрипсина можно поставить приблизительно 200 тыс. человек в США и Европе.
Проблема состоит в том, что заболевание серьезно недодиагностировано, поскольку врачи с ним зачастую незнакомы. К примеру, отчет по одному из городов США свидетельствует о проживании в нем 700 человек с PI*ZZ, но лишь 4% из них был поставлен соответствующий диагноз [3]. Недодиагностированные лица с тяжелым дефицитом ААТ, вероятно, составляют две отдельные группы: люди без клинических проявлений, несмотря на тяжелую недостаточность этого фермента, и люди с заболеваниями, но без диагноза. Относительная доля этих двух групп остается неизвестной.
КАК ЭТО РАБОТАЕТ
За последнее десятилетие значительно возросла популярность класса лекарств на основе нуклеиновых кислот, известных как РНК-лекарства. РНК-препараты — это химически модифицированные молекулы на базе РНК, которые используются для изменения свойств или функций определенных генов, транскриптов или белков. Одним из наиболее перспективных новых методов лечения с помощью РНК-препаратов является редактирование РНК, при котором синтетические олигонуклеотиды (короткие фрагменты ДНК или РНК) направляют ферменты на изменение последовательностей оснований целевых матричных РНК (мРНК) [1] [2]. Если упрощенно, редактирование оснований РНК означает переписывание генетической информации внутри неповрежденной молекулы РНК. Самые продвинутые подходы задействуют ферменты, называемые аденозиндезаминазами, действующими на РНК (ADAR) [3].
Ферменты ADAR преобразуют аденозин (A) в инозин (I) в двухцепочечных РНК. Механизм рибосомальной трансляции считывает инозин (I) как гуанозин (G), поэтому исправление патогенных точечных мутаций G-to-A видится терапевтически привлекательным. Использование синтетических олигонуклеотидов для направленного сайт-специфического ADAR-опосредованного исправления точечных мутаций имеет ряд потенциальных преимуществ, включая обратимость, независимость от клеточного цикла, избежание двухцепочечных разрывов ДНК и обход постоянных внецелевых эффектов, наблюдаемых при редактировании ДНК [4] [5].
Основной проблемой для внедрения терапевтической модальности редактирования РНК является то, что ферменты ADAR характеризуются присущей им предрасположенность к аденозинам-мишеням в определенных последовательностях, что ограничивает пространство последовательностей, которые могли бы стать мишенями [6].
WVE-006 — первый в своем классе олигонуклеотид, предназначенный для редактирования РНК непосредственно в организме человека [7].
WVE-006 исправляет мутацию одного основания в мРНК, которая закодирована Z-аллелем гена SERPINA1: путем замены основания A на I, который считывается как нужный G. Итогом становится восстановление и циркуляция функционального белка M-AAT.
WVE-006, проверенный в доклинических исследованиях, продемонстрировал мощное и продолжительное редактирование транскрипта SERPINA1 Z у мышей, восстановление уровня белка AAT до 30 мкмоль и улучшение нескольких биомаркеров заболеваний печени. WVE-006 располагает высокой специфичностью и не вызывает какого-либо побочного редактирования.
WVE-006 несет потенциал решения проблемы ассоциированных с недостаточностью альфа-1-антитрипсина заболеваний печени, легких или сразу обоих. Единый терапевтический подход позволит снизить агрегацию белка AAT в печени, восстановить циркулирующий функциональный AAT дикого типа для защиты легких от протеаз и сохранить физиологическую регуляцию белка AAT.
Подкожно вводимый WVE-006 конъюгирован с N-ацетилгалактозаминовым (GalNAc) лигандом, который связывает асиалогликопротеиновый рецептор 1 (ASGR1, ASGPR1) на гепатоцитах в целях таргетной доставки препарата. Липидные наночастицы в качестве системы доставки не используются.
КЛИНИЧЕСКИЕ ПОДРОБНОСТИ
Продолжающееся клиническое испытание RestorAATion-2 (NCT06405633) фазы Ib/IIa (нерандомизированное, открытое) оценивает безопасность, переносимость, фармакодинамику и фармакокинетику экспериментального лечения недостаточности альфа-1-антитрипсина (AATD) у взрослых пациентов с гомозиготным генотипом Pi*ZZ.
Среди основных условий включения в исследование: легко-умеренное заболевание легких, ассоциированное с AATD, что подтверждалось объемом форсированного выдоха за 1-ю секунду (ОФВ1) ≥ 50%; стабильное легкое заболевание печени, ассоциированное с AATD, что подтверждалось фиброзом на стадии, не превышающей 2-ю (≤ F2); участник не должен был курить хотя бы год до скрининга.
После того как первые 2 пациента, организм которых в принципе не способен вырабатывать собственный функциональный фермент AAT (AAT дикого типа [M-AAT]), получили одну подкожную 200-мг дозу WVE-006, уже на 3-й день начал отмечаться рост общего белка ААТ, прежде вообще неопределяемый. На 15-й день он достиг 10,8 мкмоль, что соответствует тому уровню, от которого отталкиваются регуляторы, одобряющие заместительную терапию недостаточности альфа-1-антитрипсина [1].
Тогда же циркулирующий в плазме функциональный белок М-ААТ достиг среднего уровня 6,9 мкмоль, а его вклад превысил 60% от общего количества ААТ. Рост ингибирования эластазы нейтрофилов по сравнению с исходным уровнем соответствовал выработке функционального М-ААТ.
Устойчивый синтез М-ААТ наблюдался на протяжении 57 дней.
Как ожидается, после второй дозы WVE-006 уровень М-ААТ продолжит расти.
Каких-либо претензий к профилю безопасности WVE-006 пока нет.
Результаты, во-первых, подтверждают состоятельность механизма действия WVE-006 и, во-вторых, позволяют переквалифицировать пациентов в лиц с гетерозиготным генотипом Pi*MZ (продуцирующих M-AAT хотя бы на 50-процентном уровне от нормального), у которых болезнь протекает легче и характеризуется низким риском развития заболеваний легких и печени, ассоциированных с AATD.
БИЗНЕС
В середине декабря 2022 года «Вейв лайф сайенсиз» (Wave Life Sciences) и «ГлаксоСмитКляйн» (GlaxoSmithKline) оформили четырехлетнее стратегическое соглашение о совместной разработке олигонуклеотидных препаратов. Партнерство на общую сумму максимум $3,3 млрд касается восьми лекарственных программ, за каждую из которых первая получает от второй до $130–175 млн по мере развития, плюс $200 млн по ходу продаж, плюс роялти от реализации готового препарата. Так, за WVE-006 авансом было заплачено $170 млн с обещанием выдать еще до $225 млн сообразно этапам разработки и до $300 млн в зависимости от объемов продаж, плюс роялти [1].
В 2023 году объем продаж заместительной терапии недостаточности альфа-1-антитрипсина, предназначенной только для облегчения ассоциированных заболеваний легких, составил $1,5 млрд.
«Революшн медисинс» (Revolution Medicines) готовится изменить стандарт терапии второй линии распространенного или метастатического рака поджелудочной железы.
ОСНОВНЫЕ ФАКТЫ
Экспериментальный RMC-6236 — прямой ингибитор всех основных форм онкогенных белков семейства RAS, ключевого драйвера прогрессирования протоковой аденокарциномы поджелудочной железы.
Пероральный препарат-кандидат, наделенный приемлемой переносимостью, обеспечил существенное продление жизни в сравнении с любым ныне применяемым химиотерапевтическим режимом в условиях терапии второй линии.
Осталось дождаться завершения регистрационной клинической проверки, чтобы вывести новое лечение на рынок.
«Лечение рака поджелудочной железы десятилетиями остается одной из самых незакрытых потребностей медицины. Это самый RAS-мутантный из всех основных видов рака: опухоли с мутацией RAS встречаются у более чем 90% пациентов. Продемонстрированный уровень клинической активности RMC-6236 при его дозах с приемлемой переносимостью весьма примечателен».
Брайан Уолпин (Brian Wolpin), директор Центра по изучению рака желудочно-кишечного тракта (Gastrointestinal Cancer Center) и содиректор Центра по изучению опухолей поджелудочной железы и желчевыводящей системы (Pancreas and Biliary Tumor Center) при Институте рака Дана–Фарбер (Dana–Farber Cancer Institute, DFCI, Бостон, шт. Массачусетс, США), ведущий исследователь.
«Клинические данные RMC-6236 убедительно подтвердили показатели выживаемости без прогрессирования и общей выживаемости — однозначно важные для пациентов с раком поджелудочной железы, тем самым еще ближе приблизив нас к регуляторному утверждению препарата в качестве нового стандарта лечения при распространенном или метастатическом заболевании».
Марк Голдсмит (Mark Goldsmith), исполнительный директор и председатель правления «Революшн медисинс» (Revolution Medicines).
СУТЬ ВОПРОСА
Рак поджелудочной железы (РПЖ) — одно из самых смертоносных злокачественных новообразований, характеризующееся резистентность к стандартной химиотерапии и высокой летальностью. Пятилетняя выживаемость при отдаленном метастазировании составляет незначительных 3,1% [1].
Протоковая аденокарцинома поджелудочной железы (PDAC) и ее разновидности — наиболее распространенная форма рака поджелудочной железы: на ее долю выпадает 92% всех случаев последнего. По причине отсутствия ранних симптомов и методов выявления диагноз PDAC ставится на поздней или метастатической стадии 80% пациентов.
В более чем 90% случаев опухоли PDAC несут онкогенные мутации в семействе генов RAS, которые кодируют белки — малые ГТФазы. Самой распространенной при PDAC являются мутации KRASG12X (85%), включая KRASG12D (43%) [2].
КАК ЭТО РАБОТАЕТ
RMC-6236 — пероральный мощный прямой мультиселективный ингибитор RAS-сигнализации, подавляющий активность RAS-белков как дикого типа, так и мутантных вариантов канонических RAS-изоформ (HRAS, NRAS и KRAS). RMC-6236 проявляет противоопухолевую активность в том числе при таких онкогенных RAS-мутациях, как G12X, G13X и Q61X, распространенных при протоковой аденокарциноме поджелудочной железы, немелкоклеточном раке легкого, колоректальном раке [1] [2] [3].
КЛИНИЧЕСКИЕ ПОДРОБНОСТИ
Продолжающееся клиническое исследование NCT05379985 фазы I/Ib пригласило взрослых пациентов (n=127) с ранее леченой RAS-мутантной протоковой аденокарциномой поджелудочной железы.
Среди основных исходных характеристик испытуемых: медиана возраста 64 года (30–86), мужчин 56%, медианное число предшествовавших линий терапии 2 (1–11), медианное число предшествовавших курсов лечения по метастатическому показанию 0 (1% пациентов), 1 (45%), 2+ (54%), метастазы в печени (у 67%), метастатическое заболевание на стадии IV (у 52%). Генотипическое распределение RAS-мутаций: наиболее частыми были KRASG12X (у 84%), в том числе KRASG12D (32%), KRASG12V (32%) и KRASG12X (20%).
Монотерапия при помощи RMC-6236, назначаемого ежедневно перорально в дозе от 160 до 300 мг, обеспечила следующие исходы в ходе терапии второй линии [1]:
Медиана выживаемости без прогрессирования (PFS) вышла к 8,5 месяца (95% ДИ [здесь и далее]: 5,3–11,7) и 7,6 месяца (5,9–11,1) — соответственно в популяции пациентов с мутацией KRASG12X (n=42) и любой другой RAS-мутацией (n=57).
Медиана общей выживаемости (OS) составила 14,5 месяца (8,8–NE) и 14,5 месяца (8,8–NE), притом что частота 6-месячной OS получилась равной 89% (70–97) и 91% (77–96).
Частота общего ответа (ORR) достигла 29% и 22%, тогда как частота контроля заболевания (DCR) — 91% и 89%
Применение RMC-6236 характеризовалось приемлемой переносимостью. Среди наиболее распространенных нежелательных явлений (НЯ) в ответ на лечение: сыпь (у 98% пациентов), диарея (48%), тошнота (43%), рвота (31%), стоматит (31%), усталость (20%) — они носили главным образом легко-умеренную степень тяжести. Эти НЯ вынудили 35% участников снизить дозу RMC-6236 или временно прервать лечение.
ЧТО ДАЛЬШЕ
«Революшн» продолжает трехлетнее клиническое испытание RASolute 302 (NCT06625320) фазы III, в котором RMC-6236 (ежедневно по 300 мг) сравнивается со стандартной химиотерапией во второлинейном применении среди взрослых пациентов (n=460) с протоковой адернокарциномой поджелудочной железы, прошедшей один курс лечения по метастатическому показанию. Выбор химиотерапевтического режима отдан на выбор исследователя: гемцитабин + наб-паклитаксел (GnP), оксалиплатин + лейковорин + иринотекан + 5-фторурацил (mFOLFIRINOX), липосомальный иринотекан + 5-фторурацил + лейковорин (Nal-IRI+5-FU/LV) или оксалиплатин + лейковорин + 5-фторурацил (FOLFOX). Готовность результатов ожидается к середине 2026 года.
В долгосрочных планах стоит клиническая проверка RMC-6236 в первоочередном лечении протоковой адернокарциномы поджелудочной железы. Современные химиотерапевтические подходы при этом показании выдают медиану общей выживаемости в диапазоне 8,5–11,1 месяца [1] [2] [3].
ЧТО ЕЩЕ
«Революшн» тестирует RMC-9805, пероральный селективный ковалентный ингибитор мутантного белка KRASG12D, наиболее часто встречающегося при протоковой адернокарциноме поджелудочной железы. Намечено более тщательное клиническое изучение как монотерапии RMC-9805, так и сочетанного назначения вместе с RMC-6236; подобный коктейль призван обойти потенциальную лекарственную резистентность.
ЭКСПЕРТНЫЕ КОММЕНТАРИИ
Безо всяких сомнений противоопухолевая активность RMC-6236 получилась примечательной, если отталкиваться от клинических исходов лечения протоковой аденокарциномы поджелудочной железы при помощи стандартной химиотерапии, причем независимо от ее специфики.
Так, показатели ORR, PFS и OS, зарегистрированные в ходе клинических испытаний различных химиотерапевтических режимов терапии второй линии рака поджелудочной железы укладываются в пределы 3–17%, 2,0–3,5 месяца и 6,1–6,9 месяца [1] [2] [3] [4] [5] [6] [7]. Как видим, экспериментальный RMC-6236 с лихвой их превзошел, к примеру, обеспечив более чем двукратное продление жизни.
Биотехнологический стартап «Модифай байосайенсиз» (Modifi Biosciences) разработал новый подход к лечению глиобластомы.
ОСНОВНЫЕ ФАКТЫ
Глиома — это опухоль глиальных клеток, расположенных в головном и спинном мозге. Глиобластома, наиболее распространенный тип глиомы, является крайне агрессивной опухолью мозга и остро нуждается в новых стратегиях лечения.
Глиобластому обычно лечат химиотерапевтическим препаратом темозоломидом (temozolomide), но более чем у половины пациентов развивается к нему резистентность.
«Модифай» разработала аналоги темозоломида, способные преодолевать лекарственную устойчивость глиобластомы. Экспериментальные препараты вызывают первичное повреждение ДНК, которое может быть восстановлено здоровыми клетками с неповрежденными механизмами репарации ДНК. Однако раковые клетки, лишенные таких механизмов, не в состоянии восстановить эти повреждения и со временем получают всё более токсичные вторичные повреждения, которые в свою очередь вызывают их избирательную гибель.
Планировалось, что первые клинические испытания начнутся осенью 2024 года, а в первой половине 2026-го будет организована клиническая проверка фазы II.
«Модифай», основанная в 2021 году выходцами из Йельского университета (Yale University, Нью-Хейвен, шт. Коннектикут, США), располагает инвестиционным капиталом в размере 13,1 млн долларов [1].
В конце октября 2024 года «Мерк и Ко» (Merck & Co.) купила «Модифай», выложив авансом 30 млн долларов и пообещав еще до 1,3 млрд долларов по мере развития лекарственных проектов [2].
ПРЯМАЯ РЕЧЬ
«Когда мы запустили „Модифай байосайенсиз“, то стремились радикально изменить парадигму лечения онкологических больных с глиобластомой и другими опухолями. И большая честь, что крупная фармацевтическая компания признала потенциал наших научных разработок».
Ранджит Биндра (Ranjit Bindra), соучредитель «Модифай байосайенсиз» (Modifi Biosciences), профессор терапевтической радиологии Йельской школы медицины (Yale School of Medicine, YSM, Нью-Хейвен, шт. Коннектикут, США), научный руководитель Йельского центра опухолей мозга (Yale Brain Tumor Center) при онкологической больнице Смилоу (Smilow Cancer Hospital).
«Дефекты восстановления ДНК — частая отличительная черта опухолевых клеток и одна из основных причин резистентности к противораковой терапии. Талантливая команда „Модифай байосайенсиз“ разработала новаторский подход, который, как мы считаем, располагает потенциалом лечения некоторых из наиболее рефрактерных типов рака».
Дэвид Вайнсток (David Weinstock), вице-президент по онкологическим открытиям исследовательских лабораторий «Мерк и Ко» (Merck & Co.).
КАК ЭТО РАБОТАЕТ
Генетическая нестабильность является отличительной чертой рака и обычно возникает в результате мутаций в ключевых белках репарации и/или восстановления повреждений ДНК (далее — ответ на повреждение ДНК, DDR) [1].
Подобные дефекты DDR можно использовать в терапевтических задачах при помощи ингибиторов DDR, действующих в рамках концепции синтетической летальности, которая определяется как потеря жизнеспособности в результате нарушения двух генов или сигнальных путей, которые, если они нарушены по отдельности, не являются летальными [2] [3]. Примерами успешно коммерциализированных противоопухолевых препаратов, эксплуатирующих суть синтетической летальности, являются ингибиторы поли(АДФ-рибоза)-полимераз (PARP). В любом случае избирательное уничтожение опухолевых клеток каким-либо ингибитором DDR основано или на индукции, или на сохранении повреждений ДНК или аберрантных структур ДНК.
Для того чтобы обойти механизмы лекарственной резистентности, возникающей в результате мутаций в сайте связывания DDR-белков [4], и при этом минимизировать внецелевые эффекты в здоровых клетках, располагающих достаточной экспрессией DDR, ученые «Модифай» разработали стратегию, в рамках которой лекарственное соединение модифицирует ДНК посредством двух последовательных химических этапов [5].
Первая химическая реакция направлена на создание первичного повреждения ДНК, которое быстро удаляется здоровыми DDR-профицитными клетками. Вторая химическая реакция нацелена на в 10 раз замедленное преобразование первичной модификации в более токсичное вторичное повреждение. И если скорость восстановления первичного повреждения будет достаточно быстрой, то здоровые клетки не пострадают, тогда как вторичное повреждение будет накапливаться только в ДНК опухолевых клеток с DDR-дефицитом. Этот двухступенчатый процесс позволяет преодолевать механизмы резистентности, которые снижают токсичность первичного повреждения и которые ассоциированы с устойчивостью к различным химиотерапевтическим препаратам, включая антрациклины (нарушение эксцизионной репарации нуклеозидов, NER) [6], ингибиторы топоизомеразы (потеря негомологичного соединения концов, NHEJ) [7], антиметаболиты [8] и платиносодержащие препараты (мутации в MMR) [9].
Приблизительно в половине случаев глиобластомы и более чем в 70% случаев глиомы II и III степени отсутствует O6-метилгуанин-метилтрансфераза (MGMT), белок репарации ДНК (он эпигенетически сайленсирован). Такие MGMT-дефицитные опухоли первоначально отвечают на темозоломид (temozolomide), препарат для метилирования ДНК, однако затем в половине случаев приобретают к нему лекарственную устойчивость ввиду даже незначительного ослабления активности системы репарации ошибочно спаренных оснований ДНК (MMR) [10].
Исследователи из «Модифай» придумали лекарственные соединения, которые преодолевают данный механизм резистентности, индуцируя независимое от MMR уничтожение опухолевых клеток, лишенных должной MGMT-экспрессии.
Молекулы-генотоксины, будучи своего рода высокоселективной химиотерапией, вызывают динамическое повреждение ДНК, которое могло бы быть устранено при помощи MGMT, но в условиях дефицита MGMT медленно трансформируется в межнитевую поперечную сшивку (ICL), что приводит к независимой от MMR гибели опухолевых клеток. Процесс характеризуется низкой токсичностью in vitro и in vivo, так как не затрагивает здоровые MGMT-профицитные клетки, успевающие обратить вспять первичные повреждения ДНК [11].
Экспериментальный низкомолекулярный KL-50, проверенный на животных моделях с ксенотрансплантатом резистентной к темозоломиду человеческой глиобластомы MGMT−/MMR−, продемонстрировал 7–8-кратное продление общей выживаемости в сравнении с темозоломидом и плацебо.
Лечение авторства «Модифай» подходит не только в случае рецидивирующих глиомы или глиобластомы, но и при метастатических солидных опухолях с сайленсингом MGMT, таких, к примеру, как колоректальный рак, немелкоклеточный рак легкого (НМРЛ), мелкоклеточный рак легкого (МРЛ), саркома, рак органов головы и шеи и многих других.
«Это открытие, которое приведет к невероятно значимым фармакологическим достижениям, меняет всю парадигму лечения глиомы, неизменную вот уже два десятилетия».
Роджер Штупп (Roger Stupp), руководитель отделения нейроонкологии Северо-Западного университета (Northwestern University, NU, Эванстон, шт. Иллинойс, США), содиректор Института опухолей головного мозга Мальнати (Malnati Brain Tumor Institute, MBTI, Чикаго, шт. Иллинойс, США), помощник директора Междисциплинарного онкологического центра Лури (Lurie Comprehensive Cancer Center, LCCC, Чикаго, шт. Иллинойс, США), ведущий автор основополагающих исследований, положивших начало современному лечению глиобластомы.
Фрексалимаб (frexalimab) — новый лекарственный препарат, предназначенный для лечения рецидивирующих форм рассеянного склероза.
ОСНОВНЫЕ ФАКТЫ
Фрексалимаб, разрабатываемый «Санофи» (Sanofi), обращается к совершенно иному механизму действия, чем все нынешние препараты, изменяющие течение рассеянного склероза (ПИТРС).
Фрексалимаб, таргетированный на сигнальную ось CD40–CD40L, устраняет как острое, так и хроническое нейровоспаление при рассеянном склерозе, и при этом не вызывает истощения пула B-лимфоцитов, несущее за собой риск тяжелых инфекций и иногда наблюдаемое при применении современных мощных ПИТРС, нацеленных на CD20, таких как «Окревус» (Ocrevus, окрелизумаб), «Кесимпта» / «Бонспри» (Kesimpta / Bonspri, офатумумаб) и «Бриумви» (Briumvi, ублитуксимаб).
Успешно пройдена среднестадийная клиническая проверка, продолжаются длительные регистрационные клинические испытания фазы III.
При благоприятном раскладе «Санофи» в 2027 году отправит в адрес регуляторов досье на регистрацию фрексалимаба.
ПРЯМАЯ РЕЧЬ
«По мере развития науки и диагностических инструментов менялось и наше понимание рассеянного склероза. Теперь мы точно знаем, что ингибирование CD40L приводит к сдерживанию процесса повреждения нервных клеток у людей с рассеянным склерозом. Наша вера в высокоэффективный потенциал фрексалимаба всё больше укрепляется: препарат поможет замедлить или даже остановить прогрессирование этого заболевания».
Патрик Вермерш (Patrick Vermersch), главный автор исследования, вице-президент по исследованиям в области биологии и здоровья в Университете Лилля (Université de Lille, Лилль, Франция).
«Фрексалимаб обладает уникальным механизмом действия, блокируя костимуляционный путь CD40/CD40L, который регулирует активацию и функционирование как адаптивных, так и врожденных иммунных клеток — путь, играющий ключевую роль в патогенезе рассеянного склероза. Та эффективность и скорость, с которой фрексалимаб взял заболевание под контроль, причем без истощения пула лимфоцитов, поразительна».
Гэвин Джованнони (Gavin Giovannoni), заведующий кафедры неврологии в Институте Близарда (Blizard Institute) при Школе медицины и стоматологии Бартса и Лондона (Barts and The London School of Medicine and Dentistry, Barts) в составе Лондонского университета королевы Марии (Queen Mary University of London, QMUL, Лондон, Великобритания).
«Люди с рассеянным склерозом нуждаются в новых высокоэффективных методах лечения, направленных на борьбу с прогрессированием инвалидизации — проблемой, которая по-прежнему остается одним из самых больших незакрытых медицинских вопросов. Результаты клинической проверки фрексалимаба убедительно доказали, что препарат, располагающий новым механизмом действия, способен обеспечить значимые улучшения для пациентов, страдающих этим хроническим и изнурительным заболеванием».
Эрик Вальстрём (Erik Wallström), руководитель глобального подразделения развития неврологии «Санофи» (Sanofi).
СУТЬ ВОПРОСА
CD40 и CD40L (CD154) — костимулирующий белок на поверхности антигенпрезентирующих клеток и его лиганд — играют центральную роль в регуляции гуморального и клеточно-опосредованного иммунитета: взаимодействие CD40 с CD40L на иммунных клетках участвует в активации костимулирующего сигнального пути, контролирующего «перекрестное взаимодействие» между адаптивной и врожденной иммунными системами [1].
Блокада CD40L оказалась эффективной в задаче улучшения экспериментальных аутоиммунных состояний на животных моделях, и поэтому рассматривается привлекательной терапевтической мишенью [2] [3] [4] [5] [6].
Так, на нескольких животных моделях с пептид-индуцированным аутоиммунным энцефаломиелитом (EAE) продемонстрирована функциональная роль сигнального пути CD40–CD40L: короткое профилактическое лечение антителом против CD40L (αCD40L) привело к остановке болезни [5] [7] [8] [9]. Как выяснилось, в центральной нервной системе (ЦНС) взаимодействие CD40–CD40L является критической детерминантой развития и прогрессирования заболевания [10]. Отсутствие экспрессии CD40 клетками, резидентными для ЦНС, уменьшает интенсивность и продолжительность EAE, индуцированного миелиновым олигодендроцитарным гликопротеином, и снижает степень инфильтрации воспалительных клеток в ЦНС. Энцефалитогенные Т-клетки, попадающие в ЦНС, в паренхиматозной микроглии которой CD40 отсутствует, не могут вызвать экспрессию хемокинов в ЦНС.
Клинические и патологические наблюдения за людьми c рассеянным склерозом указали на участие сигнального пути CD40–CD40L в развитии и прогрессировании заболевания [11] [12] [13] [14] [15] [16], причем с возможной связью с периферической иммунной толерантностью [17] и инфекцией вирусом Эпштейна — Барр, которая, как было уточнено, индуцирует экспрессию CD40L [18].
При рассеянном склерозе отмечается повышенный уровень экспрессии CD40L на активированных Т-клетках, что приводит к высокому уровню растворимого CD40L, и это в целом коррелирует с клиническим статусом, оцениваемым по расширенной шкале степени инвалидизации (EDSS) [12] [14] [16].
Повышенные концентрации интерферона гамма (IFNγ), обнаруженные у пациентов с рассеянным склерозом, связаны, возможно, с индуцированной CD40L усиленной выработкой интерлейкина 12 (IL-12) или, вероятно, интерлейкина 23 (IL-23) [19] [20] [21].
Выявлены генетические связи между однонуклеотидными полиморфизмами CD40 и риском развития рассеянного склероза [22] [23] [24].
Инфильтрирующие Т-клетки CD40L+ выступают в качестве движущей силы CD40-опосредованного воспалительного ответа и активируют CD40+ моноциты, макрофаги, В-клетки, эндотелиальные клетки и иммунные клетки, обитающие в ЦНС, усиливая поражение рассеянным склерозом и прогрессирование заболевания [25] [26].
Собранные данные подтверждают концепцию, что терапевтическое блокирование взаимодействия CD40–CD40L может быть эффективным подходом к лечению рассеянного склероза.
КАК ЭТО РАБОТАЕТ
Гуманизированное моноклональное IgG1-антитело фрексалимаб (frexalimab, SAR441344, INX-021) связывает CD40L, который экспрессирует на различных клетках, включая активированные T-клетки. Это приводит к ингибированию связывания CD40L с CD40, экспрессирующим на поверхности антигенпрезентирующих клеток (APC). Итогом нарушения сигнального пути CD40–CD40L становится предотвращение T-клеточно-опосредованного иммунного ответа.
Если активированные T-клетки распознают пептид, презентированный B-клетками, CD40L на T-клетках связывается с CD40 на B-клетках, приводя к активации последних. Это влечет за собой деление B-клеток, переключение изотипа антител, дифференцировку в плазматические клетки, вырабатывающие антитела против антигена-мишени. Фрексалимабу по силам сдержать этот каскад.
Выбор лиганда (CD40L), а не рецептора (CD40) в качестве мишени обусловлен рядом моментов. Во-первых, блокируется как непосредственно сигнальная ось CD40–CD40L, так и CD11, костимулирующий рецептор на APC, что приводит к сдерживанию провоспалительной поляризации цитотоксических T-клеток CD8+. Во-вторых, поляризация лимфоцитов CD4+ смещается от провоспалительной сигнализации к анергии и апоптозу T-клеток и насыщенной среде регуляторных T-клеток FoxP3+, тем самым формируя более толерогенную среду. В-третьих, CD40L экспрессирует более избирательно, чем CD40, что усиливает потенциальную безопасность и способствует фармакокинетическим, фармакодинамическим и дозировочным преимуществам.
Важно отметить, что нацеливание на сигнальную ось CD40–CD40L не приводит к истощению пула B-лимфоцитов, то есть нет рисков лимфопении — значит, нивелируются риски ослабления иммунной системы, обычно приводящего к инфекционным заболеваниям.
КЛИНИЧЕСКИЕ ПОДРОБНОСТИ
Клиническое исследование NCT04879628 фазы II (рандомизированное, двойное слепое, плацебо-контролируемое, многоцентровое, международное) пригласило взрослых (18–55 лет) пациентов (n=129) с рецидивирующими формами рассеянного склероза (RMS) —рецидивирующе-ремиттирующим рассеянным склерозом (RRMS) или вторично-прогрессирующим рассеянным склерозом (SPMS) с рецидивами.
Среди основных требований к испытуемым: либо как минимум один рецидив в течение предыдущего года, либо два и более рецидива в период двух последних лет, либо не менее одного активного очага поражения головного мозга (на МРТ-изображениях с контрастированием гадолинием) в период последних 6 месяцев до скрининга.
Среди основных характеристик участников: средний возраст 37 лет, 66% женщин, RRMS у 94%, продолжительность заболевания в среднем 7,7 года, балл по расширенной шкале степени инвалидизации (EDSS) медианных 2,5 пункта (2,0–3,5).
Пациентам назначали инъекции либо фрексалимаба в высокой дозе (1200 мг внутривенно каждые 4 недели, плюс 1800-мг нагрузочная доза в первый день) или низкой дозе (300 мг подкожно каждые 2 недели, плюс 600-мг нагрузочная доза в первый день), либо плацебо (по такой же схеме).
По прошествии 12 недель лечения число новых гиперинтенсивных очагов поражения головного мозга на T1-взвешенных МРТ-изображениях с контрастированием гадолинием, как мера активного нейровоспаления, относительно их числа на 8-й неделе составило в среднем 0,2 (95% ДИ [здесь и далее]: 0,1–0,4) и 0,3 (0,1–0,6) в группах высокой и низкой дозы фрексалимаба — против 1,4 (0,6–3,0) в объединенной группе плацебо [1] [2].
Таким образом, применение фрексалимаба снизило вероятность появления новых вышеуказанных очагов на 89% (62–97; p=0,0004) и 79% (44–92; p=0,0021) относительно плацебо: скорректированный относительный риск (risk ratio, RR) 0,11 (0,03–0,38) и 0,21 (0,08–0,56).
При этом 85% и 84% пациентов вообще не отметились появлением новых вышеобозначенных очагов — против 50% в контрольной группе.
Назначение фрексалимаба привело к снижению вероятности появления новых или разрастающихся очагов поражения головного мозга на T2-взвешенных МРТ-изображениях, как мера бремени заболевания, на 92% (74–97) и 86% (59–95): скорректированный RR 0,08 (0,03–0,26) и 0,14 (0,05–0,41). Если в группах фрексалимаба таковых было зарегистрировано 0,3 (0,1–0,6) и 0,5 (0,2–1,0), то в группе плацебо — 3,5 (1,6–7,9).
Схожая картина по сдерживанию T1- и T2-очагов сохранялась и по прошествии 24 недель лечения.
В группах фрексалимаба отмечено снижение уровня циркулирующих биомаркеров нейроаксонального повреждения и воспалительной активности — легкого полипептида нейрофиламента (NfL) и CXC-хемокина 13 (CXCL13). Плазматический уровень первого снизился на 24% и 18%, второго — на 21% и 30%, тогда как в группе плацебо, напротив, отметился подъем этих биомаркеров.
Фрексалимаб характеризовался приемлемой переносимостью. Среди наиболее распространенных нежелательных явлений (НЯ), редких и с легко-умеренной степенью тяжести: коронавирусная инфекция COVID-19 и головная боль. Тромбоэмболических событий не зафиксировано.
По завершении заслепленного периода NCT04879628 испытуемым было предложено перейти к открытому этапу исследования, когда все пациенты получают фрексалимаб, — и 97% участников (n=125/129) на это согласились.
По прошествии 36 недель (всего 48 недель с начала исследования) результаты, собранные среди оставшихся 87% людей (n=112/129), получились следующими [3] [4]:
96% и 87% пациентов, которым продолжили назначать соответственно высокую и низкую дозу фрексалимаба, были избавлены от появления новых T1-взвешенных очагов с контрастированием гадолинием.
Число таких очагов поражения оставалось низким: в среднем 0,0 и 0,2.
Число и изменение объема новых или разрастающихся T2-взвешенных очагов оставалось низким во всех группах лечения фрексалимабом.
Количество лимфоцитов было стабильным, уровни иммуноглобулина G (IgG) и иммуноглобулина M (IgM) также были стабильными или чуть снизились.
В группе высокой дозы фрексалимаба частота рецидивов в пересчете на год (ARR) были низкой, составив 0,04 (0,01–0,18), и 96% пациентов были вообще избавлены от рецидивов, чего не скажешь о группе низкой дозы, в которой ARR вышла к 0,22. Изначально, в предшествовавшем лечению году, число рецидивов было на уровне 1,3±0,6 и 1,2±0,5.
С начала исследования плазматический уровень NfL продолжал снижаться, и по окончании 48 недель его падение составило 41% и 35%.
Применение фрексалимаба характеризовалось приемлемой переносимостью. Среди наиболее распространенных НЯ: назофарингит, головная боль, ковид.
После того как прошли 72 недели (18 месяцев или 1,5 года) с начала исследования, в котором оставались 86% испытуемых (n=111/129), результаты получились следующими:
Совокупное число T1-взвешенных очагов с контрастированием гадолинием: 0,1 и 0,4 — соответственно в группах высокой и низкой дозы фрексалимаба.
Число новых или разрастающихся T2-взвешенных очагов: 0,1 и 0,4.
ARR: 0,07 (0,03–0,20) и 0,24, притом что 94% пациентов в группе высокой дозы фрексалимаба не столкнулись с рецидивом.
Усредненный балл EDSS оставался низким и стабильным.
Количество лимфоцитов и уровни иммуноглобулина почти не менялись.
Профиль безопасности фрексалимаба опасений не вызывал.
ЧТО ДАЛЬШЕ
«Санофи» продолжает клиническую проверку фрексалимаба в ходе опорных клинических исследований FREXALT (NCT06141473) и FREVIVA (NCT06141486) фазы III среди пациентов (n=1400 и n=858) соответственно с RMS и SPMS без рецидивов. Первое 3-летнее испытание, которое завершится к середине 2027 года, сравнивает фрексалимаб с «Абаджио» (Aubagio, терифлуномид), второе 4-летнее исследование, результаты которого будут готовы к концу 2026 года, — с плацебо.
ЧТО ЕЩЕ
Фрексалимаб параллельно изучается в лечении других аутоиммунных состояний — первичного синдрома Шегрена и активной системной красной волчанки: в соответствующих клинических испытаниях phaethuSA (NCT04572841) и APATURA (NCT05039840) фазы II.
Фрексалимаб проверяется в FABULINUS (NCT06111586) фазы II на предмет его способности к сохранению функции бета-клеток поджелудочной железы (эндогенной секреции инсулина) при недавно диагностированном сахарном диабете 1-го типа.
Клиническое исследование RESULT (NCT06500702) фазы II сравнивает фрексалимаб с SAR442970, нанотелом против фактора некроза опухоли (TNF) и лиганда OX40 (OX40L), и рилзабрутинибом (rilzabrutinib), обратимым ковалентным ингибитором тирозинкиназы Брутона (BTK), — в ходе лечения первичного фокально-сегментарного гломерулосклероза или первичной болезни минимальных изменений (липоидный нефроз, болезнь Нила).
ЭКСПЕРТНЫЕ КОММЕНТАРИИ
Лечение фрексалимабом рецидивирующих форм рассеянного склероза привело к благоприятным эффектам, которые отразились сдерживанием как появления новых очагов поражения головного мозга, так и разрастания существующих, а также снижением плазматических уровней релевантных биомаркеров. Всё это свидетельствует о благотворном влиянии фрексалимаба, которое проявляется торможением процессов нейроаксонального повреждения и активного нейровоспаления [1] [2] [3] [4] [5] [6].
Поисковый анализ биомаркеров подтвердил состоятельность механизма действия фрексалимаба у пациентов с рецидивирующим рассеянным склерозом: препарат оказывает мощное иммуномодулирующее действие как на врожденные, так и на адаптивные иммунные клетки без истощения лимфоцитов.
Во-первых, анализ иммунофенотипов мононуклеарных клеток периферической крови показал, что после 12 недель лечения фрексалимабом уровень плазмобластов значительно снизился, в то время как общий уровень других популяций иммунных клеток остался неизменным. Во-вторых, препарат снизил уровень циркулирующих белков, участвующих в активации и созревании В-клеток и выработке ими антител, а также белков, экспрессируемых клетками врожденного иммунитета, участвующих в дифференцировке Т-клеток и их миграции через гематоэнцефалический барьер. В-третьих, транскриптомный анализ указал на влияние фрексалимаба на хемокиновую сигнализацию, дифференцировку Т-клеток и сигнальные пути В/Т-клеточных рецепторов.
Ввиду непродолжительности и малого пациентского охвата клинического испытания фрексалимаба нельзя что-либо наверняка утверждать об улучшении клинических исходов. Так, после 12 недель терапии с рецидивом рассеянного склероза столкнулись 0% (n=0/52) и 4% (n=2/51) человек в группах высокой и низкой дозы препарата — против 4% (n=1/26) при назначении плацебо. Изменений балла по расширенной шкале степени инвалидизации (EDSS) не было ни в одной из групп. Отмечены положительные изменения по шкале влияния рассеянного склероза (MSIS-29).
Впрочем, результаты полуторагодичного применения фрексалимаба в высокой дозе, хотя и осуществленные среди очень скромной выборки из 50 пациентов, всё же позволяют сделать достаточно уверенное предположение о его высокой эффективности, если исходить из стабилизации балла EDSS и низкой среднегодовой частоте рецидивов (ARR).
Известные риски, связанные с блокированием CD40L, включают тромбоэмболические события (согласно данным о препаратах первого поколения) [7] [8], повышенную восприимчивость к инфекциям (в том числе оппортунистическим) и реакции гиперчувствительности.
В ходе применения фрексалимаба, при разработке которого был учтен опыт других молекул, несущих риск тромбоэмболических событий, таковых зарегистрировано не было.
Согласно небольшому иммунологическому исследованию торализумаба (toralizumab), моноклонального анти-CD40L-антитела первого поколения, среди пациентов с RRMS, истощения основных подмножеств лимфоцитов периферической крови не выявлено. Напротив, наблюдалось увеличение количества регуляторных Т-клеток и сдвиг в сторону противовоспалительного цитокинового ответа, что указывало на потенциальную индукцию механизмов иммунной толерантности [9].
Клиническая проверка фрексалимаба не зафиксировала истощения B-лимфоцитарного пула. В группе фрексалимаба было зарегистрировано больше инфекций по сравнению с группой плацебо, но серьезных не было. Все случаи коронавирусной инфекции COVID-19 были неосложненными, с легко-умеренной степенью тяжести.
Напротив, лечение рассеянного склероза тем же «Окревусом», моноклональным антителом против CD20, вызывающим деплецию лимфоцитов, приводит к росту риска более тяжелого течения ковида, требующего госпитализации [10], а также серьезных инфекций [11].
БИЗНЕС
«Санофи», занимающаяся фрексалимабом, лицензировала его у «Имьюнекст» (ImmuNext) в рамках соглашения, оформленного в начале января 2017 года. Французский фармацевтический гигант обязался выплатить сумму до $500 млн по мере развития лекарственного актива, плюс роялти от реализации готового препарата [1].
Впрочем, в начале мая 2024 года «Имьюнекст» продала «Роялти фарма» (Royalty Pharma), крупнейшему в отрасли бизнес-аккумулятору роялти, свои права на денежные выплаты от продаж фрексалимаба, взамен получив вознаграждение в размере $525 млн [2].
Согласно отраслевым прогнозам, фрексалимаб, если дополнительно подключит к спектру своих терапевтических показаний лечение системной красной волчанки и сахарного диабета 1-го типа, способен выйти на уровень более чем $5 млрд продаж ежегодно, из которых свыше $3 млрд будут поступать со стороны терапии рассеянного склероза.
Для «Санофи» повышенное внимание к направлению рассеянного склероза важно по ряду причин. Во-первых, «Абаджио» (Aubagio, терифлуномид) начнет терять патентную защиту после 2026 года, хотя первые дженерики, согласно договоренностям, стали появляться уже в марте 2023-го.
Так и случилось: если в период с 2020 года по 2022-й продажи «Абаджио» шли более-менее стабильно, вращаясь вокруг ежегодных €2 млрд, то в 2023 году реализация препарата упала вдвое — до €955 млн.
Во-вторых, терапевтическая эффективность терифлуномида (teriflunomide) серьезно уступает современным ПИТРС, таргетированным на CD20. Речь идет о таких лекарствах, как окрелизумаб (ocrelizumab), офатумумаб (ofatumumab) и ублитуксимаб (ublituximab).
Немецкие врачи проанализировали окрелизумаб. Его безоговорочная эффективность имеет рамки.
В-третьих, безопасность «Лемтрады» (Lemtrada, алемтузумаб) находится под большим вопросом: существуют риски серьезных сердечно-сосудистых событий, в том числе инсульта и расслоения артерий. Вот почему спрос на «Лемтраду» неуклонно падает: если в 2017 году продажи алемтузумаба (alemtuzumab) вышли к €450 млн, то в 2022-м они снизились до €80 млн, а в 2023-м «Санофи» вообще не включила в финансовый отчет сведения о реализации этого препарата.
Если «Окревус» (Ocrevus, окрелизумаб) назначать сразу после постановки диагноза рассеянного склероза, можно добиться лучших клинических результатов, чем если это делать позже — после применения других препаратов, изменяющих течение рассеянного склероза (ПИТРС).
ОСНОВНЫЕ ФАКТЫ
Раннее начало лечения рассеянного склероза «Окревусом» ассоциировано с более благоприятными клиническими исходами, нежели в случае его отложенного назначения — после иных ПИТРС.
Так, первоочередное использование окрелизумаба (ocrelizumab) привело к значительной отсрочке времени до подтвержденного ухудшения заболевания и потере способности передвигаться самостоятельно без вспомогательных приспособлений вроде трости.
Терапия первой линии рассеянного склероза окрелизумабом, если сравнивать с его применением в рамках второй и последующих линий, характеризовалась меньшим числом событий, часто ассоциирующихся с рецидивом, и сниженной вероятностью госпитализации по любой причине.
Наконец, люди, начавшие пользоваться «Окревусом», очень редко отказывались от него.
Окрелизумаб — моноклональное антитело против CD20, разработанное «Рош» (Roche) и дебютировавшее в марте 2017 года для терапии рецидивирующих форм рассеянного склероза (RMS), включая клинически изолированный синдром (CIS), рецидивирующе-ремиттирующий рассеянный склероз (RRMS), активный вторично-прогрессирующий рассеянный склероз (SPMS). Окрелизумаб также применяется в терапии первично-прогрессирующего рассеянного склероза (PPMS).
Вслед за «Окревусом» родились другие моноклональные антитела против CD20: в августе 2020 года на сцену вышел «Кесимпта» / «Бонспри» (Kesimpta / Bonspri, офатумумаб) авторства «Новартис» (Novartis), а в декабре 2022-го родился «Бриумви» (Briumvi, ублитуксимаб) идей «ТиДжи терапьютикс» (TG Therapeutics).
В России помимо окрелизумаба и офатумумаба (ofatumumab) также доступен «Ивлизи» (Ivlizi, дивозилимаб).
Все вышеуказанные препараты разрешены в терапии RMS, и только «Окревус» дозволен при PPMS, что связано, очевидно, с высокой затратностью соответствующих клинических испытаний. Однако, учитывая аналогичный механизм действия, который исповедуют лекарства конкурентов, можно смело утверждать, что данные эффективности, относящиеся к окрелизумабу, вполне применимы и к ним.
В конце июня 2024 года Европейское агентство по лекарственным средствам (EMA) и в середине сентября того же года Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) одобрили «Окревус Зуново» (Ocrevus Zunovo, окрелизумаб + гиалуронидаза) — версию окрелизумаба для подкожного введения. Новинка, которая, как и оригинальный препарат, одобрена против RMS и PPMS и назначается два раза в год, характеризуется повышенным удобством применения: 10-минутная 920-мг инъекция вместо 600-мг внутривенной инфузии длительностью не менее двух часов [1] [2].
ПРЯМАЯ РЕЧЬ
«У всех пациентов с рассеянным склерозом, независимо от его формы, болезнь прогрессирует с самого начала. Вот почему мы воодушевлены новыми данными, которые свидетельствуют, что раннее лечение „Окревусом“ приводит к существенному улучшению контроля над заболеванием, причем как при рецидивирующем, так и первично-прогрессирующем рассеянном склерозе. Обретаемый при должной терапии окрелизумабом контроль позволяет людям дольше сохранять мобильность и сдерживает ухудшение неминуемой инвалидизации».
Леви Гаррауэй (Levi Garraway), медицинский директор и глава подразделения глобального развития продукции «Рош» (Roche).
СУТЬ ВОПРОСА
Сейчас существуют две парадигмы лечения людей с рецидивирующим рассеянным склерозом [1]:
Эскалационный подход: после постановки диагноза назначается ПИТРС с низкой или умеренной эффективностью — переход к ПИТРС с высокой эффективностью осуществляется при регистрации активности заболевания или ухудшении состояния.
Ранний интенсивный подход: лечение сразу начинается с применения высокоэффективного ПИТРС, чтобы безотлагательно отсрочить ухудшение течения заболевания или добиться незамедлительного улучшения клинических исходов.
Напомним: к ПИТРС с низкой эффективностью (по степени снижения частоты рецидивов) относят интерферон бета (interferon beta), глатирамера ацетат (glatiramer acetate), терифлуномид (teriflunomide), к ПИТРС с умеренной эффективностью —диметилфумарат (dimethyl fumarate), дироксимела фумарат (diroximel fumarate), монометилфумарат (monomethyl fumarate), финголимод (fingolimod), озанимод (ozanimod), сипонимод (siponimod), понесимод (ponesimod), к ПИТРС с высокой эффективностью — натализумаб (natalizumab), алемтузумаб (alemtuzumab), митоксантрон (mitoxantrone), кладрибин (cladribine), даклизумаб (daclizumab), ритуксимаб (rituximab), окрелизумаб (ocrelizumab), офатумумаб (ofatumumab), ублитуксимаб (ublituximab).
Согласно результатам ряда исследований, ранний интенсивный подход действительно снижает число рецидивов и сдерживает ухудшение течения заболевания [1] [2] [3]. Однако отсутствовал достаточный массив данных, который бы надежно подтвердил, что указанное справедливо для окрелизумаба, если сравнивать его первоочередное назначение с отложенным применением.
БОЛЬШИЕ ЧИСЛА
«Окревус» — топовый лекарственный препарат в портфеле «Рош», ежегодно зарабатывающий для нее много денег. Вот почему швейцарский фармацевтический гигант весьма старается подчеркнуть все мыслимые преимущества окрелизумаба, которые должны стимулировать рост продаж путем наращивания и без того обширной пациентской базы: «Окревусом» пользуются более чем 350 тыс. человек по всему миру.
В период с дебютной весны 2017 года по вторую половину 2024-го включительно «Окревус» собрал для «Рош» 34,4 млрд долларов.
КЛИНИЧЕСКИЕ ПОДРОБНОСТИ
Исследование (ретроспективное, неинтервенционное, обсервационное) охватило взрослых пациентов (n=258) с рецидивирующими формами рассеянного склероза (n=198) или первично-прогрессирующим рассеянным склерозом (n=60), проходивших лечение в четырех центрах США.
Участники были разнесены на две взаимоисключающие когорты, подобранные в зависимости от начала назначения «Окревуса»: либо сразу после постановки диагноза (первая линия терапии, 1L) [n=190], либо после провала или недостаточной эффективности применения других ПИТРС (вторая или последующая линия терапии, 2L+) [n=68].
По прошествии 5 лет наблюдений выяснилось, что 25,8% пациентов (n=49/190) в когорте первоочередного применения окрелизумаба столкнулись с ухудшением течения рассеянного склероза — против 42,6% (n=29/68) в когорте его отсроченного назначения. Риск такого ухудшения среди сразу получивших лечение «Окревусом» был снижен на относительных 58%: отношение риска (hazard ratio, HR) 0,42 (95% ДИ [здесь и далее]: 0,19–0,94; p=0,03).
Конечная точка фиксировалась временем до подтвержденного ухудшения состояния, согласно шкале этапов болезни, определяемых пациентом (Patient-Determined Disease Steps, PDDS): ухудшение состояния, которое, по мнению самого пациента, отражает степень инвалидизации и которое подтверждается через 6 месяцев или позже после первого ухудшения, причем без улучшений в промежутке. Шкала PDDS имеет девять уровней — от 0 (нормальный статус) до 8 (прикованный к постели) — и предоставляет возможность оценить легкую, умеренную и выраженную степень инвалидизации.
В когорте 1L к использованию вспомогательных средств для ходьбы пришлось обратиться 6,5% пациентов (n=10/153) — против 27,4% (n=17/62) в когорте 2L+. Риск был снижен на относительных 84%: HR 0,16 (0,05–0,53; p<0,01).
Конечная точка была установлена временем до начала использования вспомогательных средств для ходьбы (трости, костыли, ходунки, коляски, трехколесные электроскутеры): подтвержденное ухудшение состояния пациента при достижении оценки PDDS < 4 баллов.
Безотлагательное после постановки диагноза назначение «Окревуса» обеспечило численно меньшую частоту рецидивов в пересчете на год (ARR) при рецидивирующе-ремиттирующем рассеянном склерозе, но без статистически значимого расхождения с когортой сравнения (p=0,73): 0,814 — против 0,863; коэффициент заболеваемости (incidence rate ratio, IRR) 1,08 (0,69–1,70).
Следует понимать, что, если сравнивать исходы применения окрелизумаба с другими ПИТРС не в рамках длительного пятилетнего срока, а только в период назначения либо «Окревуса», либо иного ПИТРС, то вышеуказанные показатели терапевтической пользы препарата «Рош» оказываются много лучше.
Предшествовавшее исследование реальной клинической практики применения окрелизумаба среди американских пациентов с рассеянным склерозом (n=694), аналогично разнесенных по двум когортам 1L (n=347) и 2L+ (n=347), сделало весьма схожие выводы [1].
Так, по прошествии 5,5 лет наблюдений в когорте 1L частота событий, часто ассоциирующихся с рецидивом (EOAR), в пересчете на год была существенно ниже, чем в когорте 2L+: 0,37 против 0,56 — разница 0,20 (0,08–0,32; p<0,001); притом что времени до наступления первого EOAR проходило существенно больше (p<0,001). Когорта 1L также засвидетельствовала существенно сниженную вероятность госпитализации по любой причине в первый год лечения: 0,021 против 0,050 (p<0,001).
Согласно другому 5-летнему исследованию применения «Окревуса» в реальной клинической практике среди итальянских пациентов с рассеянным склерозом (n=3371), первоочередное назначение окрелизумаба было ассоциировано с очень низким риском прекращения терапии по любой причине, тогда как такой же риск в случае первоочередного применения других ПИТРС был выше в 7–13 раз относительно окрелизумаба [2].
Бексикасерин (bexicaserin) — экспериментальный лекарственный препарат, предназначенный для лечения судорожных припадков по причине эпилептических энцефалопатий младенческого и детского возраста.
ОСНОВНЫЕ ФАКТЫ
«Лонгборд фармасьютикалс» (Longboard Pharmaceuticals) разработала высокоспецифическую терапию таких эпилептических состояний, тяжелых и зачастую рефрактерных к лечению, как синдром Драве (тяжелая миоклоническая эпилепсия младенчества), синдром Леннокса — Гасто, нарушение при недостаточности CDKL5, комплекс туберозного склероза и прочих.
Успешная раннестадийная клиническая проверка бексикасерина, основные результаты которой были представлены в начале января 2024 года [1], отразилась ростом биржевых котировок «Лонгборд» на более чем 300%.
Дальнейшее изучение бексикасерина подтвердило устойчивость его мощного терапевтического эффекта на фоне более чем приемлемой безопасности [2].
В середине октября 2024 года стало известно, что «Лундбек» (Lundbeck) купит «Лонгборд» за 2,6 млрд долларов наличными [3].
Если бексикасерин справится с прохождением опорной клинической программы фазы III, а затем появится на рынке, что произойдет ориентировочно к концу 2028 года, его ждет судьба бестселлера. Согласно отраслевым прогнозам, пиковые продажи бексикасерина, патентную защиту которого вполне можно продлить до 2041 года, достигнут 1,5–2 млрд долларов. При этом американский и европейский рынки лечения эпилептических энцефалопатий младенческого и детского возраста оцениваются в 6 млрд долларов к 2040 году.
ПРЯМАЯ РЕЧЬ
«Пациентские потребности в препаратах по-прежнему остаются неудовлетворенными не только при синдроме Драве, но и при многих других редких эпилептических заболеваниях. Уникальный и эффективный подход „Лонгборд“ к клинической разработке весьма примечателен, и продолжающиеся успехи бексикасерина вдохновляют как никогда».
Мэри Энн Мескис (Mary Anne Meskis), член-учредитель и исполнительный директор Фонда синдрома Драве (Dravet Syndrome Foundation, DSF).
«Устойчивый и продолжительный терапевтический ответ в виде снижения количества приступов наряду с благоприятным профилем безопасности и хорошей переносимостью среди широкого круга пациентов — всё это подтверждает потенциал бексикасерина как высокодифференцированного и лучшего в своем классе препарата. Учитывая огромную неудовлетворенную потребность пациентов, страдающих эпилептическими энцефалопатиями младенческого и детского возраста, мы намерены быстро продвигать разработку бексикасерина».
Рэндалл Кэй (Randall Kaye), медицинский директор «Лонгборд фармасьютикалс» (Longboard Pharmaceuticals).
«Я чрезвычайно горжусь командой за быстрый переход от анализа данных фазы II к запуску фазы III клинических испытаний бексикасерина, что еще раз свидетельствует о нашей всесторонней приверженности обязательствам, данным пациентам и их семьям».
Чад Оревилло (Chad Orevillo), исполнительный вице-президент и руководитель операционного отдела «Лонгборд фармасьютикалс» (Longboard Pharmaceuticals).
«„Лонгборд“ была основана для того, чтобы изменить жизнь людей, страдающих от разрушительных неврологических заболеваний. Мы многого добились, представив надежные данные с дифференцированным и инклюзивным клиническим подходом для решения проблем широкого спектра. Безграничные ресурсы „Лундбек“ ускорят реализацию нашего видения будущего, в котором люди обретут совершенно новый уровень качественной жизни».
Кевин Линд (Kevin Lind), президент и исполнительный директор «Лонгборд фармасьютикалс» (Longboard Pharmaceuticals).
«Преобразующая сделка с „Лонгборд“ ляжет краеугольным камнем в лекарственный портфель „Лундбек“ и поможет обеспечить рост бизнеса в следующем десятилетии. Благодаря приобретению бексикасерина мы продолжаем реализовывать стратегию „Сфокусированный инноватор“, улучшая жизнь пациентов, страдающих от тяжелых заболеваний головного мозга».
Карл ван Силь (Charl van Zyl), президент и исполнительный директор «Лундбек» (Lundbeck).
СУТЬ ВОПРОСА
Эпилептические энцефалопатии младенческого и детского возраста — тяжелые синдромы, ассоциированные с рефрактерными к лечению эпилептическими припадками и аномальным развитием (задержкой развития или отсутствием некоторых навыков), связанные как с этиологией основного синдрома, так и с приступами или эпилептиформными аномалиями [1] [2] [3].
В том случае, если удается улучшить контроль над приступами, то задержка в развитии может замедлиться. Однако несмотря на лечение, 40–80% пациентов всё равно сталкиваются с более чем сотней приступов каждые полгода. Заболевания обычно начинаются в раннем возрасте, зачастую в младенческом. Эпилептические припадки, как правило, остаются на всю жизнь, хотя в некоторых случаях при определенных синдромах или специфических причинах они со временем ослабевают.
Определенные области головного мозга вовлечены в различные типы приступов. Так, дисфункция контуров коры головного мозга (отмечена фиолетовым цветом), стриатума (желтым) и/или таламуса (оранжевым) является причиной абсансных и/или тонико-клонических припадков, а ствола мозга (зеленым) — индуцированные припадки. При этом поскольку последний заведует кардиореспираторной функцией, пациенты находятся в группе риска внезапной необъяснимой смерти при эпилепсии (SUDEP) [4] [5] [6].
В эту гетерогенную группу тяжелых эпилепсий входит множество заболеваний, но только для четырех из них — синдрома Драве (тяжелая миоклоническая эпилепсия младенчества), синдрома Леннокса — Гасто, нарушения при недостаточности CDKL5, комплекса туберозного склероза — есть одобренные лекарственные препараты.
Для остальных патологий этого ряда, коих насчитывается свыше двух десятков, специфического лечения пока не предложено. Речь идет о таких заболеваниях, как синдром Ангельмана, синдром Ландау — Клеффнера, синдром Отахара, синдром Уэста, ранняя миоклоническая энцефалопатия, эпилепсия с миоклонически-астатическими приступами, эпилептическая энцефалопатия с непрерывными спайками и волнами во время сна, связанные с генетическими мутациями эпилепсии (затрагивают гены SCN2A, SCN8A, KCNQ2, KCNQ3, KCNT1, SynGAP1) и др.
КАК ЭТО РАБОТАЕТ
Доклинические и клинические данные подтверждают роль рецепторов 5-гидрокситриптамина (5-HT; серотониновые рецепторы) подсемейства 2 (5-HT2) в модуляции частоты и порога возникновения судорожных припадков [1] [2] [3] [4].
Агонизм серотонинового рецептора подтипа 2C (5-HT2C) приводит к стимулированию высвобождения гамма-аминомасляной кислоты (ГАМК), что отражается повышением порога возбудимости нейронов и снижает вероятность возникновения эпилептических приступов. Агонисты 5-HT2C-рецептора являются эффективными средствами лечения различных моторных судорог и судорожных расстройств.
Модуляция рецепторов 5-HT2C в гиппокампальных пирамидальных ГАМК-эргических нейронах подавляет их повышенную возбудимость. У мышей с нокаутом рецепторов 5-HT2C наблюдаются спонтанные судороги и фиксируется сниженный порог для проконвульсивных стимулов. Мета-хлорфенилпиперазин (mCPP) повышает порог для миоклонических и тонических судорог, индуцированных пентилентетразолом и электрошоком; эффект блокируется антагонистом 5-HT2C. В генетической модели синдрома Драве агонист 5-HT2C ослабил судорогоподобное поведение и эпилептиформную электрическую активность у мутантных данио-рерио scn1Lab−/− [5] [6] [7] [8] [9] [10] [11] [12].
Бексикасерин (bexicaserin, LP352) — пероральный низкомолекулярный суперагонист 5-HT2C центрального действия, специально разработанный для применения при эпилептических энцефалопатиях младенческого и детского возраста.
Бексикасерин характеризуется более чем 200-кратной избирательностью в отношении лиганд-связывающего участка рецептора 5-HT2C, если сравнивать с рецепторами 5-HT2A и 5-HT2B, и это минимизирует нежелательные явления (НЯ), отмечаемые при агонизме рецепторов 5-HT2A и 5-HT2B.
Напомним: разработка агонистов рецептора 5-HT2C всегда была непростой задачей ввиду серьезных НЯ, вызванных отсутствием селективности относительно агонизма рецепторов 5-HT2A и 5-HT2B. Так, активация рецептора 5-HT2A вызывает галлюцинации, бессонницу, эйфорию и тревожность, а активация рецептора 5-HT2B ассоциирована с нарушением функций клапанов сердца и, возможно, легочной артериальной гипертензией (ЛАГ) [13] [14].
Ранее на рынок поступили такие агонисты 5-HT2C-рецептора, как фенфлурамин (fenfluramine) и лоркасерин (lorcaserin), однако их далеко не безопасный профиль ввиду неселективности действия привел, во-первых, к запрету применения обоих препаратов для лечения ожирения (первый характеризовался кардиотоксичностью, второй был ассоциирован с развитием рака) и, во-вторых, к ограничению использования фенфлурамина только в терапии синдрома Драве и синдрома Леннокса — Гасто.
Лоркасерин попал под запрет ввиду опасности развития раковых заболеваний.
Первые клинические исследования бексикасерина продемонстрировали обнадеживающие результаты [15] [16]:
бексикасерин быстро поступал в системную циркуляцию после перорального приема;
не наблюдалось клинически значимого влияния пищи на системную экспозицию бексикасерина;
концентрация пролактина после приема бексикасерина транзиторно повышалась дозозависимым образом, что указывает на успешное подключение центральных 5-HT2C-рецепторов;
вызванные лечением НЯ в основном носили легко-умеренную степень тяжести: головная боль, сонливость, головокружение (в том числе постуральное), учащенное мочеиспускание и ортостатическая гипотензия.
«Арена нейросайенсиз» (Arena Neuroscience), запущенная в январе 2020 года как самостоятельное подразделение «Арена фармасьютикалс» (Arena Pharmaceuticals) и в октябре того же года сменившая название на «Лонгборд фармасьютикалс» (Longboard Pharmaceuticals), разработала бексикасерин с прицелом на куда большую избирательность к 5-HT2C-рецептору в сравнении с лоркасерином.
КЛИНИЧЕСКИЕ ПОДРОБНОСТИ
Клиническое исследование PACIFIC (NCT05364021) фазы Ib/IIa (рандомизированное, двойное слепое, плацебо-контролируемое, многоцентровое) пригласило пациентов (n=52) в возрасте 12–65 лет (из США и Австралии) с диагнозом синдрома Драве, синдрома Леннокса — Гасто или другой эпилептической энцефалопатии младенческого и детского возраста, включая комплекс туберозного склероза, нарушение при недостаточности CDKL5, связанные с SCN2A-мутацией эпилепсии и пр.
Среди основных требований к участию: минимум 4 моторных приступа в месяц на фоне приема 1–4 противосудорожных лекарственных препаратов.
Испытуемым назначали перорально плацебо или бексикасерин три раза в день в дозе 6, 9 или 12 мг. В течение первых 15 дней проводилось постепенное повышение дозы до максимально переносимой. Затем осуществлялось поддерживающее лечение на протяжении 60 дней. Было разрешено продолжать придерживаться курса противосудорожных препаратов, таких, к примеру, как клобазам (clobazam), вальпроат (valproat), каннабидиол (cannabidiol), ламотриджин (lamotrigine), леветирацетам (levetiracetam).
Применение бексикасерина обеспечило снижение частоты моторных приступов за 28-дневный период наблюдений на медианных 53% — против снижения на 21% в группе плацебо. Скорректированная на плацебо разница в их частоте составила 33% [1].
Если говорить об отдельных заболеваниях, то при синдроме Драве, синдроме Леннокса — Гасто или другой эпилептической энцефалопатии младенческого и детского возраста медианное уменьшение частоты моторных приступов вышло к 72%, 48% и 61% соответственно. Относительно плацебо показатели разницы в подгруппах пациентов с синдромом Леннокса — Гасто или другим эпилептическим заболеванием получились равными 27% и 29%.
Среди наиболее распространенных нежелательных явлений (НЯ) при назначении бексикасерина: сонливость, снижение аппетита, запор, диарея, летаргия, тремор, инфекции мочевыводящих путей, усталость, пирексия, ажитация. Вызванные лечением НЯ вынудили 21% пациентов прекратить прием бексикасерина.
Анализ собранных данных за весь цикл лечения выявил повышенную эффективность бексикасерина, которому удалось снизить частоту моторных приступов на медианных 60% — против снижения на 17% в группе плацебо: разница составила 42% (p=0,0538). Однако, согласно эксплораторному анализу post hoc, скорректированная на плацебо частота моторных приступов уменьшилась благодаря бексикасерину на усредненных 52% (p=0,0206). При синдроме Драве, синдроме Леннокса — Гасто или другой эпилептической энцефалопатии младенческого и детского возраста медианное снижение частоты моторных приступов получилось равным 75%, 51% и 66%.
Назначение бексикасерина привело к снижению частоты всех (не только моторных) приступов на 51% — против снижения на 21% в группе плацебо.
При этом пропорция респондентов (снижение частоты приступов не менее чем на 50%) составила 60% и 51% в случае моторных и любых эпилептических приступов соответственно — против 33% и 33% в контрольной группе.
Следует отметить примечательную переносимость бексикасерина: в поддерживающем периоде терапии максимальную 12-мг дозу препарата получали 77% испытуемых.
Согласно промежуточным 9-месячным данным продолжающегося 52-недельного открытого клинического исследования NCT05626634 фазы II, которое охватило пациентов, завершивших испытание PACIFIC, но пожелавших продолжить экспериментальное лечение бексикасерином (n=41), число моторных приступов оставалось сниженным на медианных 58% с момента начала участия в последнем, тем самым свидетельствуя об устойчивом терапевтическом эффекте [2].
Профиль безопасности бексикасерина характеризовался приемлемой переносимостью, хотя один человек вышел из исследования ввиду побочного эффекта, проявившегося вялостью и усталостью, а еще два пациента отказались от терапии по иным причинам, не связанным с лечением. Среди наиболее распространенных НЯ: инфекции верхних дыхательных путей, COVID-19, синусит, снижение аппетита, пирексия, снижение веса.
ЧТО ДАЛЬШЕ
В конце сентября 2024 года «Лонгборд» запустила клиническое испытание DEEp SEA фазы III, которое изучит способность бексикасерина сдерживать эпилептические приступы при синдроме Драве у пациентов (n=160) в возрасте 2 лет и старше [1].
Вскоре будет дан старт еще одному клиническому исследованию фазы III, DEEp OCEAN, которое проверит бексикасерин среди пациентов (n=320) с синдромом Леннокса — Гасто и другими эпилептическими энцефалопатиями младенческого и детского возраста.
«Лонгборд» также протестирует усовершенствованную рецептуру бексикасерина для назначения дважды в день, а не трижды.
ЭКСПЕРТНЫЕ КОММЕНТАРИИ
Если сравнивать терапевтическую эффективность бексикасерина (bexicaserin) с одобренными противосудорожными лекарственными препаратами, такими как «Финтепла» (Fintepla, фенфлурамин), «Эпидиолекс» (Epidiolex, каннабидиол) и «Зталми» (Ztalmy, ганаксолон), то в задаче уменьшения числа приступов при эпилептических энцефалопатиях младенческого и детского возраста экспериментальное лекарство выглядит весьма конкурентоспособным и даже превосходящим.
Более того, бексикасерин потенциально подходит для применения при рефрактерных к лечению эпилептических состояний любой этиологии.
Профиль безопасности бексикасерина существенно лучше, чем таковой у фенфлурамина (fenfluramine), который очень эффективен, но несет риски серьезных сердечно-сосудистых проблем.
Пациентам с эпилептическими энцефалопатиями приходится принимать сразу множество различных противосудорожных препаратов, которые ингибируют и/или индуцируют ферменты CYP (цитохромы P450), что приводит к клинически значимым лекарственным взаимодействиям. Бексикасерин прекрасно справляется со своей терапевтической задачей даже в условиях сложной полифармакотерапии. Так, CYP2D6 и CYP3A4 однозначно не влияют на его метаболизм, а почечные транспортеры или P-гликопротеин (P-gp) вряд ли в нем участвуют, притом что непосредственно метаболизм этой молекулы осуществляется через УДФ-глюкуронилтрансферазу (UGT) с образованием глюкуронидного метаболита M20.
Согласно долгосрочным наблюдениям, лечение меланомы при помощи препарата «Китруда» (Keytruda, пембролизумаб) авторства «Мерк и Ко» (Merck & Co.) оказалось успешнее, чем лекарством «Ервой» (Yervoy, ипилимумаб) разработки «Бристол-Майерс Сквибб» (Bristol-Myers Squibb).
«Прогноз для пациентов с диагнозом меланома непрестанно улучшается: смертность снизилась на 30% по сравнению с десятилетней давностью. Поразительно, но свыше трети больных, прошедших терапию „Китрудой“, живы и сегодня, спустя десять лет после лечения».
Каролин Робер (Caroline Robert), руководитель отделения дерматологии в онкологической клинике Гюстава Русси (Вильжюиф, Франция) и содиректор подразделения исследований меланомы при Университете Париж-юг (Орсе, Франция).
КЛИНИЧЕСКИЕ ПОДРОБНОСТИ
В клиническом исследовании KEYNOTE-006 (NCT01866319) фазы III осуществлялось сравнение эффективности и безопасности лечения распространенной или неоперабельной меланомы (на стадии III или IV) PD-1-блокатором пембролизумабом (pembrolizumab) или CTLA-4-блокатором ипилимумабом (ipilimumab).
Пациенты проходили терапию тем или иным иммуноонкологическим препаратом либо до завершения испытания (максимальный срок 2 года), либо до момента прогрессирования заболевания, неприемлемой токсичности или нежелания участника продолжать.
Если пациент демонстрировал полный ответ (CR), подтвержденный двумя сканированиями с интервалом не менее 4 недель, он считался излечившимся. Пациенты, достигшие стабилизации заболевания (SD) или лучшего статуса во время первого курса пембролизумаба, могли пройти второй его курс (на срок до 1 года).
На основе результатов, собранных в KEYNOTE-006, регулятор в лице Управления по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) согласовал дебют «Китруды», первого в мире блокатора PD-1, состоявшийся в начале сентября 2014 года.
«Мерк и Ко» пошла дальше: по завершении KEYNOTE-006 участникам предложили продолжить наблюдаться в рамках исследования KEYNOTE-587 (NCT03486873) фазы III, основной задачей которого было установить, как долго излечившиеся будут оставаться в статусе ремиссии и, если их болезнь вернется, сколько они проживут, получая новое лечение пембролизумабом (вышеупомянутый второй курс).
По итогам наблюдений, продолжавшихся медианных 123,7 месяца (122,0–127,3) от момента начала KEYNOTE-006, вероятность остаться в живых на протяжении 10 лет после лечения распространенной меланомы пембролизумабом составила 34,0% — против 23,6% после лечения ипилимумабом [1] [2].
Медиана общей выживаемости (OS) вышла к 32,7 месяца (95% ДИ [здесь и далее]: 24,5–41,6) — против 15,9 месяца (13,3–22,0): отношение риска (hazard ratio, HR) 0,71 (0,60–0,85).
Медиана выживаемости без прогрессирования (PFS) получилась равной 9,4 месяца (6,7–11,6) — против 3,8 месяца (2,9–4,3): HR 0,64 (0,54–0,75).
Особый интерес представляют те больные, которые прошли терапию «Китрудой» на протяжении не менее чем 94 недель. Среди этих участников медиана OS достигнута не была, тогда как 8-летняя вероятность сохранить жизнь составила 80,8%.
«Десять лет назад препарат „Китруда“, ставший первой анти-PD-1/L1-терапией, создал основу для революционных прорывов в лечении меланомы и других видов рака. Пембролизумаб кардинальным образом изменил подход к лечению солидных опухолей. Мы со все возрастающим нетерпением ждем очередных инноваций для пациентов».
Марджори Грин (Marjorie Green), старший вице-президент и руководитель направления онкологии подразделения глобального клинического развития исследовательских лабораторий «Мерк и Ко» (Merck & Co.).
СУТЬ
Долгосрочные результаты наблюдений безоговорочно подтвердили статус пембролизумаба как стандарта лечения неоперабельной меланомы.
«Джемперли» (Jemperli, достарлимаб) существенно расширил спектр показаний: отныне его можно назначать всем пациентам с первичным распространенным или рецидивирующим раком эндометрия.
ОСНОВНЫЕ ФАКТЫ
Соответствующее разрешение выдано Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) в начале августа 2024 года [1].
«Джемперли» назначается вначале совместно с химиотерапевтическими карбоплатином и паклитакселом, а затем самостоятельно.
Ранее «Джемперли» был дозволен для первоочередного лечения первичного распространенного или рецидивирующего рака эндометрия с дефектами в системе репарации ошибочно спаренных оснований ДНК (dMMR) или высокочастотной микросателлитной нестабильностью (MSI-H) [2].
До этого достарлимаб (dostarlimab), блокатор PD-1, за которым стоят «ГлаксоСмитКляйн» (GlaxoSmithKline) и «Анаптисбайо» (AnaptysBio), применялся на более поздних этапах лечения: для терапии распространенного или рецидивирующего рака эндометрия с dMMR, который либо прогрессировал во время или после предшествовавшей платиносодержащей терапии, либо когда пациент не подходит для хирургического вмешательства или облучения [3].
Вообще же «Джемперли», который дебютировал в конце августа 2021 года [4], разрешено универсально использовать в лечении любых рецидивирующих или распространенных солидных опухолей с dMMR, прогрессировавших во время или после терапии — в том случае, если нет удовлетворительных альтернативных вариантов лечения [5].
По состоянию на начало сентября 2024 года, в России достарлимаб не зарегистрирован.
Достарлимаб поможет при любых солидных опухолях с MSI-H/dMMR.
ПРЯМАЯ РЕЧЬ
«„Джемперли“ предложил сообществу больных раком эндометрия столь необходимую новую схему лечения, которая дает надежду жить дольше и даже выжить».
Эдриенн Мур (Adrienne Moore), выжившая пациентка, член-основатель и президент Афроамериканской сети по борьбе с раком эндометрия (ECANA).
«Добавление достарлимаба к стандартным химиопрепаратам доказало привнесение существенной пользы при раке эндометрия во всей популяции пациентов».
Мэтью Пауэлл (Matthew Powell), руководитель отделения гинекологической онкологии в Медицинской школе Вашингтонского университета (Сент-Луис, шт. Миссури, США).
«„Джемперли“ с химиотерапией — первая и единственная иммуноонкологическая схема лечения, которая продемонстрировала продление общей выживаемости при неоперабельном раке эндометрия вне зависимости от статуса биомаркеров и которая смогла охватить большую выборку пациентов, прежде лишенных эффективных вариантов терапии».
Хешам Абдулла (Hesham Abdullah), старший вице-президент и руководитель глобальных онкологических исследований и разработок «ГлаксоСмитКляйн» (GlaxoSmithKline).
КЛИНИЧЕСКИЕ ПОДРОБНОСТИ
Клиническое исследование RUBY (NCT03981796) фазы III (рандомизированное, двойное слепое, плацебо-контролируемое, многоцентровое, международное) пригласило взрослых женщин с рецивирующим или распространенным раком эндометрия.
Заболевание участников должно было быть либо на стадии III или IV, либо в статусе первого рецидива с низкой вероятностью успеха хирургии и/или облучения. Пациенты либо не должны были вообще ранее проходить системную противораковую терапию, либо должны были столкнуться с рецидивом или прогрессированием заболевания по прошествии не менее чем 6 месяцев после неоадъювантной или адъювантной таковой.
Испытуемым назначали либо достарлимаб, карбоплатин и паклитаксел, а затем только достарлимаб, либо плацебо, карбоплатин и паклитаксел, а затем только плацебо. Применение достарлимаба осуществлялось до момента прогрессирования заболевания или неприемлемой токсичности, на протяжении максимум 3 лет.
Оценка эффективности лечения проводилась в когорте пациентов с dMMR/MSI-H.
По прошествии наблюдений на протяжении медианных 24,8 (19,2–36,9) месяца результаты, обеспеченные применением достарлимаба, получились следующими [1]:
снижение риска прогрессирования или смерти на 72%: медиана выживаемости без прогрессирования (PFS) составила 30,3 (95% ДИ [здесь и далее]: 11,8–NR) месяца — против 7,7 (5,6–9,7) месяца в группе контроля, отношение риска (hazard ratio, HR) 0,28 (0,16–0,50), p<0,0001;
вероятность сохранения статуса PFS на протяжении не менее чем 24 месяцев: 61% (46–73) — против 16% (7–27);
вероятность общей выживаемости (OS) на протяжении как минимум 24 месяцев: 83% (67–92) — против 59% (43–71), HR 0,30 (0,13–0,70);
частота общего ответа (ORR): 74% (58–86), включая 26% полных ответов (CR) и 48% частичных ответов (PR), — против 62% (47–76), в том числе CR 11% и PR 51%;
длительность ответа (DoR) не вышла (NR) к медианному значению (3,4–28,3+) — против 5,4 (2,7–27,2+) месяца.
После наблюдений на протяжении медианных 36,6 (31,0–48,7) месяца результаты назначения достарлимаба следующие [2] [3]:
медиана OS не достигнута (NE–NE) — против 31,4 (20,3–NE) месяца, HR 0,32 (0,17–0,63; p=0,0002);
вероятность остаться в живых сроком 2 и 3 года составила 83% (70–91) и 78% (64–87) — против 58% (44–69) и 46% (33–58).
Если говорить о пациентах без статуса dMMR/MSI-H, то есть без дефектов в системе репарации ошибочно спаренных оснований ДНК (pMMR) или с микросателлитной стабильностью (MSS), то после наблюдений на протяжении медианных 37,5 (31,2–49,5) месяца результаты применения достарлимаба следующие:
медиана OS вышла к 34,4 (28,6–NE) месяца — против 27,0 (21,5–35,6) месяца, HR 0,79 (0,60–1,04; p=0,0493);
вероятность остаться в живых сроком 2 и 3 года составила 67% (59–73) и 49% (41–56) — против 53% (46–60) и 42% (34–49).
Сводные данные для всей популяции пациентов (без учета статуса dMMR/MSI-H), за которыми наблюдали в течение медианных 37,2 (31,0–49,5) месяца, таковы:
медиана OS получилась равной 44,6 (32,6–NE) месяца — против 28,2 (22,1–35,6) месяца, HR 0,69 (0,54–0,89; p=0,002);
вероятность остаться в живых на протяжении 2 и 3 лет составила 70% (64–76) и 55% (48–61) — против 54% (48–60) и 43% (36–49).
«Джемперли» обеспечил полную ремиссию ректальной аденокарциномы.
ОДНАКО
В середине июня 2024 года «Китруда» (Keytruda, пембролизумаб), блокатор PD-1 авторства «Мерк и Ко» (Merck & Co.), добавил 40-е по счету онкологическое показание, подключив точно такое же, как «Джемперли», лечение первичного распространенного или рецидивирующего рака эндометрия [1].
В клиническом исследовании KEYNOTE-868 (NCT03914612) фазы III, пембролизумаб (pembrolizumab), вначале назначаемый вместе с карбоплатином и паклитакселом, а затем самостоятельно, снизил риск прогрессирования заболевания или смерти на 70% и 40% при dMMR и pMMR: HR 0,30 (0,19–0,48; p<0,0001) и 0,60 (0,46–0,78; p<0,0001) [2].
Другими словами, эффективность «Китруды» сопоставима с таковой у «Джемперли».
ЭКСПЕРТНЫЕ КОММЕНТАРИИ
Рак эндометрия занимает шестое место по распространенности среди женщин во всём мире и является вторым по частоте видом гинекологического рака [1] [2] [3] [4] [5].
Карбоплатин c паклитакселом — стандартная химиотерапевтическая схема в рамках первой линии лечения первичного распространенного или рецидивирующего рака эндометрия, однако исходы остаются неудовлетворительными: медиана общей выживаемости не превышает 3 лет [6] [7] [8] [9].
Солидные опухоли, характеризующиеся дефектами в системе репарации ошибочно спаренных оснований ДНК (dMMR) или высокочастотной микросателлитной нестабильностью (MSI-H), встречаются в 25–30% случаев рака эндометрия [10] [11] [12]. Повышенная экспрессия рецептора программируемой клеточной смерти 1 (PD-1) и его лигандов (PD-L1 и PD-L2), а также высокая мутационная нагрузка опухоли (TMB), ассоциированная с dMMR/MSI-H, делают такие опухоли потенциально восприимчивыми к терапии, направленной против PD-1 или PD-L1 [10] [13] [14].
Достарлимаб (dostarlimab) — ингибитор иммунных контрольных точек (ИИКТ), направленный против PD-1, что приводит к блокированию связывания последнего со своими лигандами с последующей реактивацией противоопухолевого иммунитета.
Тот факт, что достарлимаб работает и в случае солидных опухолей с отсутствием дефектов в системе репарации ошибочно спаренных оснований ДНК (pMMR) или микросателлитной стабильностью (MSS), хотя частота ответа и его глубина меньше, нежели при dMMR/MSI-H, обусловлен тем, что пусть уровень TMB относительно небольшой, зато экспрессия PD-L1 присутствует почти всегда [15].
Назначение достарлимаба на фоне цитотоксической химиотерапии связано с тем, что последняя оказывает иммуномодулирующее действие, в том числе вмешиваясь в иммуносупрессивные пути и усиливая цитотоксический Т-клеточный ответ [16]. Комбинированное лечение из химиотерапии и иммунотерапии синергически влияет на микроокружение опухоли [17] [18] [19] [20] [21]. При использовании подобной комбинации против нескольких видов рака были продемонстрированы клинические преимущества, включая улучшение выживаемости [22] [23] [24] [25] [26] [27] [28].
Что примечательно, эффективность лечения достарлимабом не отметилась единообразием во всех предопределенных подгруппах пациентов. Так, при раке эндометрия на III стадии или при отсутствии признаков заболевания иммунотерапия проявила себя незначительно, а местами даже хуже, чем стандартное лечение. Впоследствии, впрочем, когда собралось больше данных, наметилась тенденция к улучшению исходов. Не исключено, это связано с ограниченным объемом выборки таких пациентов и относительно коротким периодом наблюдений, потому что нет никаких биологических предпосылок, чтобы они отвечали иначе [7] [29] [30] [31] [32].
Хотя клиническое испытание RUBY (NCT03981796) доказало пользу применения достарлимаба с химиопрепаратами при раке эндометрия, всё еще неизвестно, как поведет себя мононазначение достарлимаба. Продолжающиеся клинические исследования DOMENICA (NCT05201547) и KEYNOTE-C93 (NCT05173987) пытаются восполнить этот пробел: они проверяют первоочередное использование соответственно достарлимаба и пембролизумаба в сравнении с карбоплатином и паклитакселом при распространенном или рецидивирующем раке эндометрия [33] [34].
Приличное продление общей выживаемости, обеспеченное иммунотерапией, затрагивает актуальный вопрос об оптимальной ее продолжительности. Выбор максимум трехлетнего назначения достарлимаба был обусловлен тем, что средняя продолжительность жизни пациентов с первичным распространенным или рецидивирующим раком эндометрия на стандартной химиотерапии не превышала этого срока. Опять же, в RUBY, как и в других испытаниях иммуноонкологического лечения, наблюдалось плато кривой выживаемости без прогрессирования.
«Интарсия терапьютикс» (Intarcia Therapeutics) разработала ITCA 650 — подкожный имплантат, предназначенный для длительной поддерживающей терапии сахарного диабета 2-го типа.
ITCA 650 представляет собой миниатюрную осмотическую помпу. Крошечное устройство подкожно имплантируется в брюшную стенку, а затем до шести месяцев кряду организует непрерывную доставку в организм микродоз эксенатида (exenatide), агониста рецептора глюкагоноподобного пептида 1 (GLP1RA).
После установки мини-помпы можно фактически на полгода забыть о том, что необходимо постоянно принимать противодиабетические лекарственные препараты.
Польза неоспорима: считается, что до 70% диабетиков в какой-то момент времени прекращают хроническую терапию изнуряющего заболевания. Во многом это объясняется тем тяжелым бременем, которое налагают обязательства каждодневного контроля над уровнем глюкозы в крови.
Всё бы ничего, но Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA), начиная с 2017 года, планомерно отклоняет заявки «Интарсия» на регистрацию ITCA 650.
ТЕХНИЧЕСКИЕ МОМЕНТЫ
Осмотическая мини-помпа ITCA 650, которой занимается «Интарсия», представляет собой медицинское изделие, заключенное в цилиндрический корпус-резервуар из титанового сплава размером со спичку (44×4 мм). После того как в амбулаторных условиях девайс, работающий без каких-либо электронных компонентов, подкожно установлен в брюшную стенку, начинается непрерывная доставка в организм микродоз эксенатида.
Благодаря солевому градиенту внеклеточная жидкость поступает через полупроницаемую мембрану в осмотическое устройство мини-помпы ITCA 650. Образующееся давление очень медленно и c постоянным усилием толкает поршень, воздействующий на емкость с лекарственным соединением, которое высвобождается из отверстия диффузора.
Разработка технологии лекарственной доставки Medici Drug Delivery System отняла у специалистов «Интарсия» долгих десять лет: пришлось решать вопросы с оптимизацией рецептуры эксенатида, чтобы его действующее вещество, на которое воздействует температура тела, не только оставалось стабильным как минимум год, но и при этом сохраняло эффективность процесса микродозирования [1].
Впрочем, основную часть усилий по практическому воплощению непосредственного конструкции ITCA 650 взяла на себя «Алза» (Alza), в марте 2000 года выпустившая «Виадур» (Viadur) — подкожно имплантируемую во внутреннюю часть плеча титановую мини-помпу, сделанную по фирменной технологии доставки DUROS и на протяжении года высвобождавшую лейпрорелин (leuprorelin), также известный как лейпролид (leuprolide), синтетический пептидный аналог гонадотропин-рилизинг-гормона (GnRH). «Виадур» предназначалась для паллиативной терапии распространенного рака предстательной железы [2] [3].
Коммерциализацией «Виадура» занималась «Байер» (Bayer) — вплоть до конца 2007 года, когда было принято решение прекратить реализацию мини-помпы ввиду изменившихся рыночных условий [4] [5]. В 2001 году «Алза» была поглощена «Джонсон энд Джонсон» (Johnson & Johnson) за $10,5 млрд [6] [7]. В августе 2006 года «Интарсия» лицензировала у «Джонсон энд Джонсон» технологию доставки DUROS.
Удобство мини-помы ITCA 650 для пациентов очевидно: существующие рецептуры эксенатида — «Баета» (Byetta) и «Бидуреон» / «Баета Лонг» (Bydureon / Byetta Long) авторства «АстраЗенека» (AstraZeneca) — требуют подкожного инъекционного введения дважды в день и один раз в неделю соответственно.
Да и всё прочее множество GLP1RA-препаратов, таких, к примеру, как «Виктоза» (Victoza, лираглутид), «Адликсин» / «Ликсумия» (Adlyxin / Lyxumia, ликсисенатид), «Танзеум» / «Эперзан» (Tanzeum / Eperzan, албиглутид), «Трулисити» (Trulicity, дулаглутид) и «Оземпик» (Ozempic, семаглутид), представлено в инъекционной форме.
Особняком стоит «Ребелсас» (Rybelsus, семаглутид) — первый в мире GLP1RA-препарат в пероральном таблеточном исполнении, разработанный «Ново Нордиск» (Novo Nordisk) и дебютировавший в конце сентября 2019 года.
Novo Nordisk выпустила семаглутид в виде таблеток.
Согласно ряду исследований, менее половины пациентов с сахарным диабетом 2-го типа выходят к целевому уровню глюкозы в крови. Неоптимальный гликемический контроль влечет за собой повышенную заболеваемость и смертность, необходимость заниматься лечением диабетических осложнений, рост расходов системы здравоохранения. В большинстве случаев это объясняется равно как низкой осведомленностью и информированием больных и недостаточным уровнем их доходов, так и сложностью или неудобством противодиабетической терапии [8] [9].
ЭФФЕКТИВНОСТЬ И БЕЗОПАСНОСТЬ
Эффективность и безопасность ITCA 650 были подтверждены в опорной клинической программе FREEDOM из четырех испытаний фазы III (рандомизированных, двойных слепых или открытых, плацебо-контролируемых или с активным контролем, многоцентровых, международных) среди взрослых пациентов с сахарным диабетом 2-го типа:
FREEDOM-1 (NCT01455857): пациенты (n=450) с уровнем гликированного гемоглобина (HbA1c) в диапазоне 7,5–10%, придерживающиеся диеты и физических упражнений и принимающие метформин, производные сульфонилмочевины или тиазолидинедион (либо их комбинации), не способные достичь должного гликемического контроля. Сравнение с плацебо.
FREEDOM-1 HBL (NCT01785771): вышеуказанная популяция пациентов (n=75), но с высоким HbA1c (>10,0%). Сравнение с плацебо.
FREEDOM-2 (NCT01455870): пациенты (n=500) с HbA1c в диапазоне 7,5–10,5%, принимающие метформин и не способные достичь должного гликемического контроля. Сравнение с «Янувией» (Januvia, ситаглиптин), ингибитором дипептидилпептидазы-4 (DPP-4).
FREEDOM-CVO (NCT01455896): пациенты (n=4000) с HbA1c > 6,5% и сердечно-сосудистым заболеванием в анамнезе (коронарным, цереброваскулярным или периферических артерий) или факторами риска развития таковых. Сравнение с плацебо.
Мини-помпа ITCA 650 продемонстрировала статистически значимое превосходство над плацебо и другими противодиабетическими препаратами сравнения в том, что касается снижения уровня HbA1c, веса и риска серьезных сердечно-сосудистых событий (MACE), таких как кардиоваскулярная смерть, нелетальный инфаркт миокарда, нелетальный инсульт или госпитализация по причине нестабильной стенокардии [1] [2] [3] [4] [5].
Какой из препаратов сильнее снижает вес: «Оземпик» и «Вегови» или «Мунджаро» и «Зепбаунд»?
РЕГУЛЯТОРНЫЕ ОТКАЗЫ
В феврале 2017 года «Интарсия» отправила в FDA регистрационное досье на противодиабетическую мини-помпу ITCA 650, контролируемым образом высвобождающую эксенатид.
В сентябре регулятор отказал, сославшись на недочеты в проведенных клинических испытаниях и производственные неполадки, повлиявшие на качество готового изделия.
В феврале 2018 года стало известно о проблемах в отношении долгосрочных стабильности и стерильности мини-помпы.
В сентябре 2019 года «Интарсия» отослала заявку повторно [1]. В марте 2020 года FDA во второй раз отклонило регистрационное досье ITCA 650 [2].
В ЧЕМ ПРОБЛЕМА
По мнению экспертов FDA, установка мини-помпы ITCA 650 ассоциирована с ростом риска острого повреждения почек (ОПП), иногда приводящего к длительной госпитализации с сопутствующими осложнениями, такими как необходимость в диализе или смертельный исход [1].
С серьезным нежелательным явлением (НЯ) в лице ОПП столкнулись 14 пациентов (0,6%) с установленным имплантатом ITCA 650 — против 4 человек (0,2%), получивших плацебо. Что примечательно, риск развития ОПП оказался выше, если сравнивать с практикой применения уже одобренных GLP1RA-препаратов. Более того, если в случае последних события ОПП вскрылись лишь в ходе постмаркетинговых наблюдений, то при использовании ITCA 650 они были зарегистрированы во время проведения клинических испытаний, адекватных и строго контролируемых.
Большинство ОПП, протекавших в серьезной форме, сопровождались рвотой, диареей и обезвоживанием — известными НЯ в ответ на терапию эксенатидом. Это подтверждает причинно-следственную связь между установкой ITCA 650 и развитием ОПП.
Предложенные «Интарсия» меры по снижению риска острого повреждения почек не являются оптимальными, поскольку серьезные случаи ОПП произошли у испытуемых без известных факторов риска, таких как почечная недостаточность средней и тяжелой степени или прием определенных лекарственных препаратов.
Как указали специалисты американского регулятора, «Интарсия» не предоставила надежных доказательств, что действие мини-помпы ITCA 650 не связано с чрезмерным сердечно-сосудистым риском.
Метаанализ данных, собранных в клинических испытаниях FREEDOM-1 (NCT01455857), FREEDOM-2 (NCT01455870) и FREEDOM-CVO (NCT01455896), выяснил, что установка мини-помпы ITCA 650 сопровождалась 12-процентным ростом риска серьезных сердечно-сосудистых событий (MACE), таких как кардиоваскулярная смерть, нелетальный инфаркт миокарда, нелетальный инсульт или госпитализация по причине нестабильной стенокардии: отношение риска (hazard ratio, HR) 1,12 (95% ДИ [здесь и далее]: 0,83–1,51). Указанный риск особенно себя проявил в подгруппе возрастных участников (65 лет и старше): HR 1,67 (1,02–2,71). Напротив, применение утвержденных GLP1RA-препаратов характеризуется снижением сердечно-сосудистых рисков.
Кроме того, вскрылись проблемные места, связанные с конструктивными особенностями мини-помпы ITCA 650.
Пристальное изучение фармакологических характеристик ITCA 650 in vitro, обусловленных спецификой ее конструкции, не смогло подтвердить, что заявленные параметры высвобождения лекарственных средств соответствуют предполагаемому медицинскому применению мини-помпы in vivo. Отмечено непостоянство в доставляемой дозировке в течение дня, то есть невозможно утверждать, что ITCA 650 уверенно справится с задачей контролируемого высвобождения. Анализ отказов ITCA 650 в работе не позволил безоговорочно утверждать о достаточной надежности изделия для долгосрочной эксплуатации.
Инспекция предприятия «Интарсия» выявила недочеты в практике производства мини-помпы ITCA 650.
Невозможно подтвердить безоговорочную стерильность ITCA 650, если исходить из оценок, связанных с целостностью упаковочно-укупорочной системы для промежуточного хранения компонентов устройства, контактирующим с изделием оборудованием для розлива, процессом депирогенизации компонентов первичной упаковочно-укупорочной системы, методикой проверки на эндотоксины. Нельзя исключать риск выпуска ITCA 650, лишенных действующего вещества или микробиологически контаминированных.
Более чем разочарованные инвесторы «Интарсия», которые уже, очевидно, устали ждать положительных исходов, сдаваться, впрочем, не собирались. Предприятие продолжило прилагать максимум усилий, чтобы решить все проблемные вопросы и добиться благосклонности регулятора.
К чему изобретать своё, если можно взять готовое? А ты, Eli Lilly, страшись!
ЧТО БЫЛО ДАЛЬШЕ
Когда-то инвесторы профинансировали деятельность основанной в 1997 году «Интарсия» на $1,7 млрд, оценочная стоимость частной компании превышала $5,5 млрд, а коммерческие прогнозы будущего мини-помпы ITCA 650 витали на оптимистичном уровне многих миллиардов долларов [1] [2].
«Интарсия» также вынашивала планы выпуска усовершенствованной версии осмотической мини-помпы, способной контролируемо высвобождать лекарственный препарат на протяжении целого года.
После категорического нежелания FDA включать новаторскому устройству зеленый свет десятки сотрудников «Интарсия» уволились, включая генерального директора, штаб-квартира в Бостоне закрылась, а корпоративный сайт начал перенаправлять на веб-ресурс другой компании — «Ай-ту-оу терапьютикс» (i2o Therapeutics).
Пришлось свернуть дополнительные клинические испытания фазы III, в том числе сравнивающие ITCA 650 с «Джардинсом» (Jardiance, эмпаглифлозин), ингибитором натрий-глюкозного контранспортера 2-го типа (SGLT-2), или глимепиридом (glimepiride), производным сульфонилмочевины, среди придерживающихся терапии метформином пациентов (NCT03060980).
В конце августа 2023 стало известно, что активы «Интарсия» отошли в руки «Ай-ту-оу» — биотехнологического стартапа, в который было вложено венчурных $90 млн и который был запущен в 2019 году с целью вывода на рынок кардиометаболических лекарственных средств длительного действия: GLP1RA i2o-105s, агониста рецептора амилина i2o-107, двойного агониста инкретиновых гормонов i2o-120, агониста рецептора глюкагона i2o-130 [3] [4].
В руках предприятия находится фирменная платформа ионных жидкостей, лицензированная у лаборатории Самира Митраготри (Samir Mitragotri) из Гарвардского университета и предназначенная для производства биологических препаратов и пептидов в пероральном исполнении [5].
«Ай-ту-оу» возглавил Курт Грейвс (Kurt Graves), руководитель «Интарсия», а заведовать наукой стал Эндрю Юнг (Andrew Young), аналогично пришедший из «Интарсия».
Трехлетний курс лечения тирзепатидом против лишнего веса на 94% снизил риск развития сахарного диабета 2-го типа.
ТОЧКА
Несмотря на затянувшуюся патовую ситуацию, надежда на успешное будущее ITCA 650 всё же оставалась.
В феврале 2023 года FDA, после продолжительных дебатов с «Интарсия», всё же согласилось провести общественные слушания при участии консультативного комитета по эндокринологическим и метаболическим препаратам, чтобы окончательно разрешить все спорные моменты.
В конце сентября 2023 года искомые слушания состоялись. Что интересно, регулятор отклонил просьбу о предоставлении «Интарсия» нескольких часов, обычно выделяемых для презентации своего проекта экспертному совету [1] [2] [3].
В предваряющих заседание комментариях Курт Грейвс напомнил экспертам, что FDA одобрило множество GLP1RA-препаратов, включая «Оземпик» (Ozempic, семаглутид) и «Вегови» (Wegovy, семаглутид), несмотря на риск острого повреждения почек: «Это центральный вопрос для всего класса GLP1RA, и потому следует признать существование такого риска, а также проблем с ЖКТ и обезвоживанием, а затем сделать всё необходимо, чтобы улучшить профиль пользы и безопасности. Учитывая ошеломляющий рост спроса на GLP1RA-препараты, это крайне важно» [4].
По итогам консультативный комитет единогласно (19 голосов против, 0 голосов за) пришел к выводу о нецелесообразности одобрения ITCA 650, так как ее преимущества не перешивают риски [5].
В середине августа 2024 года FDA вынесло вердикт, положивший конец многолетним усилиям «Интарсия»: мини-помпа ITCA 650 одобрена не будет, апелляцию подавать запрещено [6].
ИЗ ИСТОРИИ
В ноябре 2014 года «Интарсия» передала «Лабораториям Сервье» (Servier Laboratories) права на реализацию ITCA 650 за пределами США и Японии — за авансовых $171 млн; с потенциалом совокупных выплат на сумму свыше $1 млрд [1].
В марте 2015 года «Интарсия» договорилась со швейцарской «Ньюмаб терапьютикс» (Numab Therapeutics) о совместном изучении ITCA 650 в применимости мини-помпы для доставки в организм фрагментов антител: одноцепочечного ND017 и триспецифичного ND016 — соответственно для терапии метаболических расстройств (диабета, ожирения) и аутоиммунных заболеваний [2]. ND016, составленный из трех высокостабильных вариабельных доменов, одновременно блокирует два провоспалительных цитокина, интерлейкин 17A (IL-17A) и фактор некроза опухоли (TNF), и дополнительно связывается с сывороточным альбумином [3].
В сентябре 2015 года «Интарсия» купила основанный выходцами из «ГлаксоСмитКляйн» (GlaxoSmithKline) биотехнологический стартап «Фаундри фармасьютикалс» (Phoundry Pharmaceuticals) с портфелем оптимизированных пептидов, надеясь, что ITCA 650 пригодится для их доставки в ходе лечения диабета и ожирения [4].
Идеи «Интарсия» заразили Фонд Билла и Мелинды Гейтс, который в конце 2016 года пообещал вложить в компанию до $140 млн, чтобы та разработала мини-помпу для доконтактной профилактики ВИЧ-инфицирования (PrEP). Речь идет о непрерывной доставке препятствующего распространению вируса по организму лекарственного соединения: например, «Трувады» (Truvada, тенофовир + эмтрицитабин) или каботегравира (cabotegravir).
В январе 2017 года «Интарсия» подружилась с Калифорнийским институтом биомедицинских исследований (Calibr) при научно-исследовательском институте «Скриппс рисёрч» (Scripps Research): на предмет улучшения фармакологических свойств (потентности и времени полувыведения) эксенатида в составе ITCA 650 — путем включения связывающего мотива сывороточного белка в ковалентную боковую цепь эксенатида в целях усиления его спиральности [5] [6].
ТЕМ ВРЕМЕНЕМ
Со всех сторон привлекательная концепция подкожно имплантируемой мини-помпы ITCA 650 заставила биотехнологический стартап «Вивани медикал» (Vivani Medical) заняться реализацией проекта схожей направленности.
Экспертиза «Вивани» обращается к технологии NanoPortal, лежащей в основе биосовместимых, миниатюрных, подкожных имплантатов, предназначенных для доставки лекарственных молекул, включая пептидные, на протяжении длительного периода времени. Имплантаты, не имеющие ни электроники, ни движущихся частей, обращаются к нанопористой мембране из оксида титана, состоящей из миллионов нанотрубок с точным размером, внутренний диаметр которых заранее отрегулирован в соответствии с размером молекул лекарственного вещества.
Установка имплантата снимает ряд критических вопросов терапии. Во-первых, низкая приверженность лечению ввиду необходимости хронического приема препарата в нужное время и в нужной дозе влечет за собой потенциальные перерывы в терапии, что нежелательным образом сказывается на течении заболевания. Во-вторых, колебания лекарственной концентрации в организме по причине особенностей профиля высвобождения могут отражаться нежелательными явлениями.
«Вивани», осведомленная о проблемах «Интарсия», полагает, что причиной провала ITCA 650 является нерегулярное и неконтролируемое высвобождение эксенатида, следствием которого были нежелательные явления критического для FDA характера. Предприятие уверено, что решения на базе NanoPortal характеризуются минимальными колебаниями профиля высвобождения. Дополнительным преимуществом является вдвое меньший размер имплантата (2,2×21,5 мм против 4×45 мм), что позволяет устанавливать его менее травмирующим образом.
Экспериментальный имплантат NPM-115, будучи основным проектом «Вивани», ориентирован на хроническую коррекцию лишнего веса при помощи высокодозного эксенатида, высвобождаемого на протяжении шести месяцев.
NPM-119 идентичен NPM-115, разве что доза эксенатида снижена, поскольку имплантат предназначен для терапии сахарного диабета 2-го типа.
NPM-139 содержит семаглутид, который высвобождается в течение целого года и который должен помочь в лечении ожирения.
Особняком стоит OKV-119 — разрабатываемый в партнерстве с «Окава фармасьютикалс» (Okava Pharmaceuticals) имплантат для лечения преддиабета, ожирения и диабета у кошек.
Согласно доклиническим исследованиям на мышиных моделях с ожирением, NPM-115 обеспечил снижение веса приблизительно на 20% по сравнению с плацебо по прошествии 28 дней, что соответствует эффективности еженедельных инъекций семаглутида.
На четвертый квартал 2024 года намечен запуск клинического исследования LIBERATE-1, которое проверит терапевтическую состоятельность и безопасность NPM-115 среди лиц (n=24), страдающих ожирением или избыточной массой тела. Вначале всем участникам будут назначаться еженедельные инъекции семаглутида на протяжении восьми недель. После рандомизации одной группе будет установлен имплантат, тогда как две контрольные группы в течение девяти недель будут получать еженедельные инъекции семаглутида или эксенатида [1].